
Evaluation of a Live Attenuated S. sonnei Vaccine Strain in the Human Enteroid Model

 ,
,
Abstract
1. Introduction
2. Results
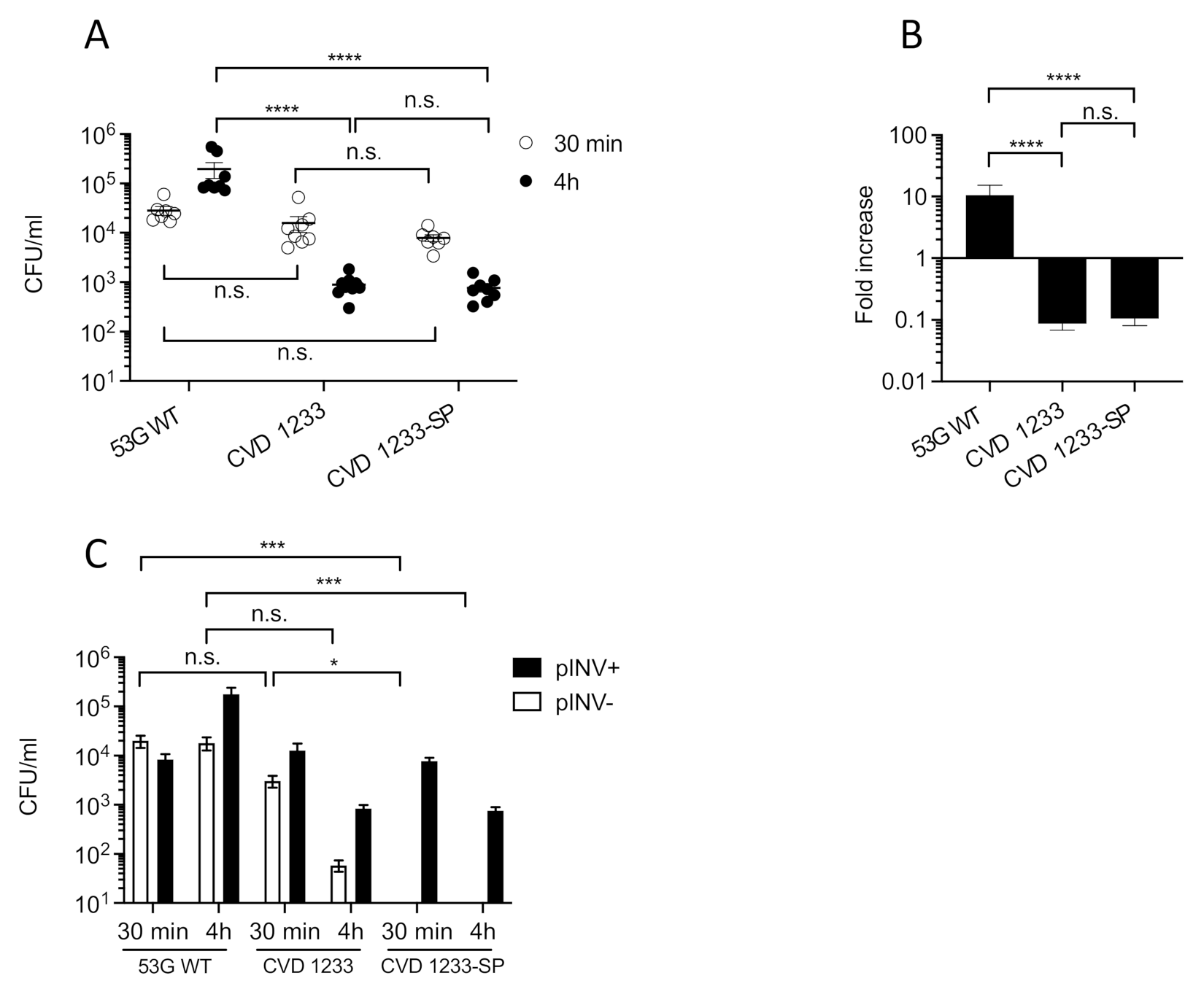
2.1. Increased Stabilization of pINV Improves the Consistency of S. sonnei Vaccine Strain Production
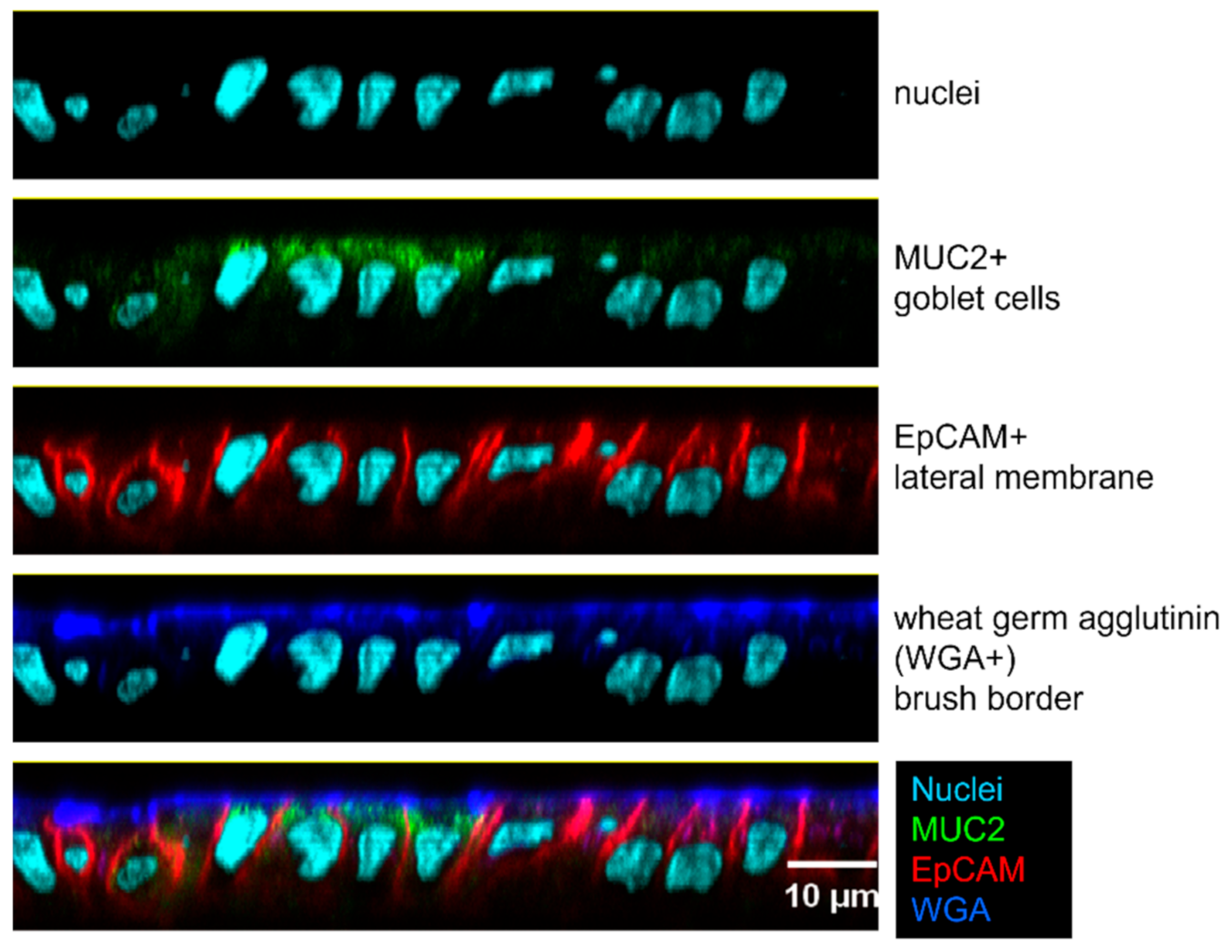
2.2. Use of a Human Enteroid Model to Validate CVD 1233-SP
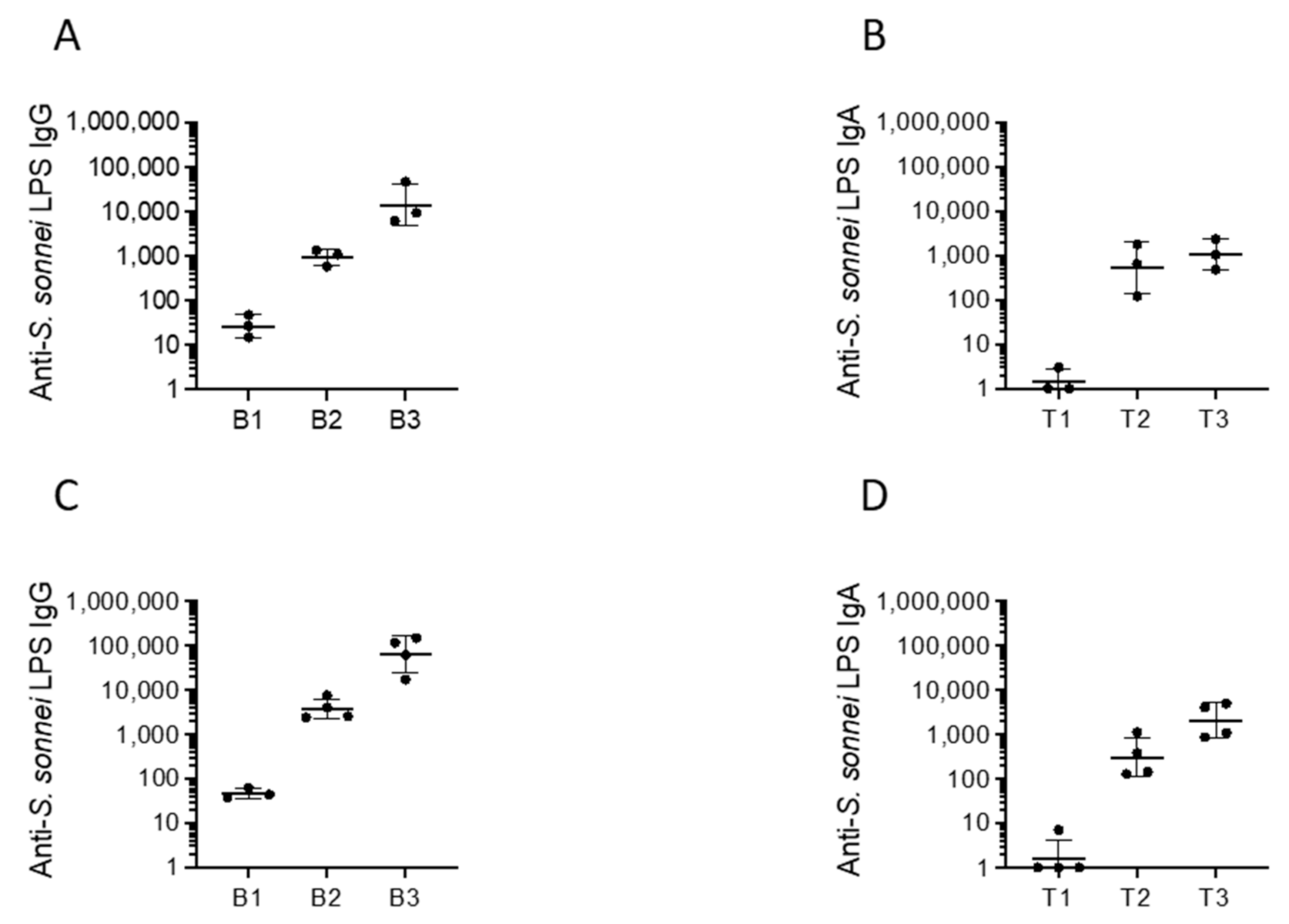
2.3. Immunogenicity, Protectoin, and Safety of CVD 1233-SP in Guinea Pigs
2.4. Use of CVD 1233-SP as a Live Vector for Expression of Heterologous Antigens from ETEC
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Construction of Strains
4.3. Congo Red Loss Assay
4.4. HT29 Cell Cultivation, Invasion Assays, and Adherence Inhibition Assays
4.5. Enteroid Cultivation and Invasion Assays
4.6. Immunizations, Sample Collection and Challenge
4.7. ELISA
4.8. Enteroid Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Baker, S.; The, H.C. Recent insights into Shigella. Curr. Opin. Infect. Dis. 2018, 31, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Tickell, K.D.; Brander, R.L.; Atlas, H.E.; Pernica, J.M.; Walson, J.L.; Pavlinac, P.B. Identification and management of Shigella infection in children with diarrhoea: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1235–e1248. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Liu, J.; Platts-Mills, J.A.; Juma, J.; Kabir, F.; Nkeze, J.; Okoi, C.; Operario, D.J.; Uddin, J.; Ahmed, S.; Alonso, P.L.; et al. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: A reanalysis of the GEMS case-control study. Lancet 2016, 388, 1291–1301. [Google Scholar] [CrossRef]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Livio, S.; Strockbine, N.A.; Panchalingam, S.; Tennant, S.M.; Barry, E.M.; Marohn, M.E.; Antonio, M.; Hossain, A.; Mandomando, I.; Ochieng, J.B.; et al. Shigella isolates from the global enteric multicenter study inform vaccine development. Clin. Infect. Dis. 2014, 59, 933–941. [Google Scholar] [CrossRef]
- Thompson, C.N.; Duy, P.T.; Baker, S. The Rising Dominance of Shigella sonnei: An Intercontinental Shift in the Etiology of Bacillary Dysentery. PLoS Negl. Trop. Dis. 2015, 9, e0003708. [Google Scholar] [CrossRef]
- Ud-Din, A.I.; Wahid, S.U.; Latif, H.A.; Shahnaij, M.; Akter, M.; Azmi, I.J.; Hasan, T.N.; Ahmed, D.; Hossain, M.A.; Faruque, A.S.; et al. Changing trends in the prevalence of Shigella species: Emergence of multi-drug resistant Shigella sonnei biotype g in Bangladesh. PLoS ONE 2013, 8, e82601. [Google Scholar] [CrossRef]
- Vinh, H.; Nhu, N.T.; Nga, T.V.; Duy, P.T.; Campbell, J.I.; Hoang, N.V.; Boni, M.F.; My, P.V.; Parry, C.; Nga, T.T.; et al. A changing picture of shigellosis in southern Vietnam: Shifting species dominance, antimicrobial susceptibility and clinical presentation. BMC Infect. Dis. 2009, 9, 204. [Google Scholar] [CrossRef]
- Collins, J.P.; Friedman, C.R.; Birhane, M.G.; Karp, B.E.; Osinski, A.; Montgomery, M.W.; Thomas, D.; Barkley, J.; Sanchez, M.C.; Hanna, S.; et al. Evidence of Failure of Oral Third-Generation Cephalosporin Treatment for Shigella sonnei Infection. Open Forum. Infect. Dis. 2020, 7, ofaa113. [Google Scholar] [CrossRef]
- Baker, K.S.; Dallman, T.J.; Field, N.; Childs, T.; Mitchell, H.; Day, M.; Weill, F.X.; Lefevre, S.; Tourdjman, M.; Hughes, G.; et al. Horizontal antimicrobial resistance transfer drives epidemics of multiple Shigella species. Nat. Commun. 2018, 9, 1462. [Google Scholar] [CrossRef]
- Barry, E.M.; Pasetti, M.F.; Sztein, M.B.; Fasano, A.; Kotloff, K.L.; Levine, M.M. Progress and pitfalls in Shigella vaccine research. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 245–255. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Riddle, M.S.; Platts-Mills, J.A.; Pavlinac, P.; Zaidi, A.K.M. Shigellosis. Lancet 2018, 391, 801–812. [Google Scholar] [CrossRef]
- Mani, S.; Wierzba, T.; Walker, R.I. Status of vaccine research and development for Shigella. Vaccine 2016, 34, 2887–2894. [Google Scholar] [CrossRef]
- Riddle, M.S.; Chen, W.H.; Kirkwood, C.D.; MacLennan, C.A. Update on vaccines for enteric pathogens. Clin. Microbiol. Infect. 2018, 24, 1039–1045. [Google Scholar] [CrossRef]
- Walker, R.I.; Wierzba, T.F.; Mani, S.; Bourgeois, A.L. Vaccines against Shigella and enterotoxigenic Escherichia coli: A summary of the 2016 VASE Conference. Vaccine 2017, 35, 6775–6782. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.M.; Wang, J.; Wu, T.; Davis, T.; Levine, M.M. Immunogenicity of multivalent Shigella-ETEC candidate vaccine strains in a guinea pig model. Vaccine 2006, 24, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- McVicker, G.; Tang, C.M. Deletion of toxin-antitoxin systems in the evolution of Shigella sonnei as a host-adapted pathogen. Nat. Microbiol. 2016, 2, 16204. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Doucet, M.; Grassel, C.L.; Delaine-Elias, B.; Zachos, N.C.; Barry, E.M. Evaluating Shigella flexneri Pathogenesis in the Human Enteroid Model. Infect. Immun. 2019, 87, e00740-18. [Google Scholar] [CrossRef]
- Koestler, B.J.; Ward, C.M.; Fisher, C.R.; Rajan, A.; Maresso, A.W.; Payne, S.M. Human Intestinal Enteroids as a Model System of Shigella Pathogenesis. Infect. Immun. 2019, 87, e00733-18. [Google Scholar] [CrossRef] [PubMed]
- Pilla, G.; McVicker, G.; Tang, C.M. Genetic plasticity of the Shigella virulence plasmid is mediated by intra- and inter-molecular events between insertion sequences. PLoS Genet. 2017, 13, e1007014. [Google Scholar] [CrossRef] [PubMed]
- Foulke-Abel, J.; In, J.; Kovbasnjuk, O.; Zachos, N.C.; Ettayebi, K.; Blutt, S.E.; Hyser, J.M.; Zeng, X.L.; Crawford, S.E.; Broughman, J.R.; et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp. Biol. Med. 2014, 239, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Foulke-Abel, J.; In, J.; Yin, J.; Zachos, N.C.; Kovbasnjuk, O.; Estes, M.K.; de Jonge, H.; Donowitz, M. Human Enteroids as a Model of Upper Small Intestinal Ion Transport Physiology and Pathophysiology. Gastroenterology 2016, 150, 638–649.e8. [Google Scholar] [CrossRef]
- In, J.; Foulke-Abel, J.; Zachos, N.C.; Hansen, A.M.; Kaper, J.B.; Bernstein, H.D.; Halushka, M.; Blutt, S.; Estes, M.K.; Donowitz, M.; et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 48–62.e3. [Google Scholar] [CrossRef]
- Noel, G.; Baetz, N.W.; Staab, J.F.; Donowitz, M.; Kovbasnjuk, O.; Pasetti, M.F.; Zachos, N.C. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. 2017, 7, 45270. [Google Scholar] [CrossRef]
- Zachos, N.C.; Kovbasnjuk, O.; Foulke-Abel, J.; In, J.; Blutt, S.E.; de Jonge, H.R.; Estes, M.K.; Donowitz, M. Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J. Biol. Chem 2016, 291, 3759–3766. [Google Scholar] [CrossRef]
- Middendorp, S.; Schneeberger, K.; Wiegerinck, C.L.; Mokry, M.; Akkerman, R.D.; van Wijngaarden, S.; Clevers, H.; Nieuwenhuis, E.E. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 2014, 32, 1083–1091. [Google Scholar] [CrossRef]
- Barry, E.M.; Levine, M.M. A tale of two bacterial enteropathogens and one multivalent vaccine. Cell Microbiol. 2019, 21, e13067. [Google Scholar] [CrossRef]
- Wu, T.; Grassel, C.; Levine, M.M.; Barry, E.M. Live attenuated Shigella dysenteriae type 1 vaccine strains overexpressing shiga toxin B subunit. Infect. Immun. 2011, 79, 4912–4922. [Google Scholar] [CrossRef][Green Version]
- McKenzie, G.J.; Craig, N.L. Fast, easy and efficient: Site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 2006, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Poole, S.T.; Maciel, M., Jr.; Dinadayala, P.; Dori, K.E.; McVeigh, A.L.; Liu, Y.; Barry, E.; Grassel, C.; Prouty, M.G.; Renauld-Mongenie, G.; et al. Biochemical and Immunological Evaluation of Recombinant CS6-Derived Subunit Enterotoxigenic Escherichia coli Vaccine Candidates. Infect. Immun. 2019, 87, e00788-18. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Smith, E.M.; Foulke-Abel, J.D.; Barry, E.M. Research in a time of enteroids and organoids: How the human gut model has transformed the study of enteric bacterial pathogens. Gut Microbes 2020, 12, 1795492. [Google Scholar] [CrossRef] [PubMed]
- Lemme-Dumit, J.M.; Doucet, M.; Zachos, N.C.; Pasetti, M.F. Epithelial and neutrophil interactions and innate immune response to Shigella in a human intestinal enteroid-neutrophil co-culture. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chanin, R.B.; Nickerson, K.P.; Llanos-Chea, A.; Sistrunk, J.R.; Rasko, D.A.; Kumar, D.K.V.; de la Parra, J.; Auclair, J.R.; Ding, J.; Li, K.; et al. Shigella flexneri Adherence Factor Expression in In Vivo-Like Conditions. mSphere 2019, 4, e00751-19. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Blutt, S.E.; Ettayebi, K.; Zeng, X.L.; Broughman, J.R.; Crawford, S.E.; Karandikar, U.C.; Sastri, N.P.; Conner, M.E.; Opekun, A.R.; et al. Human Intestinal Enteroids: A New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J. Virol. 2016, 90, 43–56. [Google Scholar] [CrossRef]
- Llanos-Chea, A.; Citorik, R.J.; Nickerson, K.P.; Ingano, L.; Serena, G.; Senger, S.; Lu, T.K.; Fasano, A.; Faherty, C.S. Bacteriophage Therapy Testing Against Shigella flexneri in a Novel Human Intestinal Organoid-Derived Infection Model. J. Pediatr Gastroenterol. Nutr. 2019, 68, 509–516. [Google Scholar] [CrossRef]
- Donowitz, M.; Turner, J.R.; Verkman, A.S.; Zachos, N.C. Current and potential future applications of human stem cell models in drug development. J. Clin. Invest. 2020, 130, 3342–3344. [Google Scholar] [CrossRef]
- Hartman, A.B.; Venkatesan, M.M. Construction of a stable attenuated Shigella sonnei DeltavirG vaccine strain, WRSS1, and protective efficacy and immunogenicity in the guinea pig keratoconjunctivitis model. Infect. Immun. 1998, 66, 4572–4576. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Taylor, D.N.; Sztein, M.B.; Wasserman, S.S.; Losonsky, G.A.; Nataro, J.P.; Venkatesan, M.; Hartman, A.; Picking, W.D.; Katz, D.E.; et al. Phase I evaluation of delta virG Shigella sonnei live, attenuated, oral vaccine strain WRSS1 in healthy adults. Infect. Immun. 2002, 70, 2016–2021. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barnoy, S.; Baqar, S.; Kaminski, R.W.; Collins, T.; Nemelka, K.; Hale, T.L.; Ranallo, R.T.; Venkatesan, M.M. Shigella sonnei vaccine candidates WRSs2 and WRSs3 are as immunogenic as WRSS1, a clinically tested vaccine candidate, in a primate model of infection. Vaccine 2011, 29, 6371–6378. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Fonseka, S.; Boren, T.; Ranallo, R.T.; Suvarnapunya, A.E.; Lee, J.E.; Barnoy, S.; Venkatesan, M.M. Further characterization of Shigella sonnei live vaccine candidates WRSs2 and WRSs3-plasmid composition, invasion assays and Sereny reactions. Gut Microbes 2011, 2, 244–251. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Altboum, Z.; Barry, E.M.; Losonsky, G.; Galen, J.E.; Levine, M.M. Attenuated Shigella flexneri 2a Delta guaBA strain CVD 1204 expressing enterotoxigenic Escherichia coli (ETEC) CS2 and CS3 fimbriae as a live mucosal vaccine against Shigella and ETEC infection. Infect. Immun. 2001, 69, 3150–3158. [Google Scholar] [CrossRef] [PubMed]
- Kopecko, D.J.; Washington, O.; Formal, S.B. Genetic and physical evidence for plasmid control of Shigella sonnei form I cell surface antigen. Infect. Immun. 1980, 29, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad Sci USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | Attack Rate | Efficacy |
|---|---|---|
| CVD 1233 § | 1/7 | 86% |
| CVD 1233-SP | 0/4 | 100% |
| Unvaccinated controls § | 6/6 | - |
| CVD 1233-SP::CS2-CS3 | 1/5 | 80% |
| Unvaccinated controls | 3/3 | - |
| Strain | Description | Reference |
|---|---|---|
| 53G WT | Wild-type S. sonnei 53G | [45] |
| 53G-SP | 53G WT with stabilized pINV | This work |
| CVD 1233 | ΔguaBA, Δsen, S. sonnei vaccine derived from 53G WT | [17] |
| CVD 1233-SP | CVD 1233 with stabilized pINV | This work |
| CVD 1233::CS2-CS3 | CVD 1233-expressing CS2 and CS3 antigens from ETEC | This work |
| CVD 1233-SP::CS2-CS3 | CVD 1233::CS2-CS3 with stabilized pINV | This work |
| Primer | Sequence (5′-3′) | Generated Strains |
|---|---|---|
| Wu215_F | ACCGAACAACGAACTGTTGGAA | CVD 1233::CS2-CS3 and CVD 1233-SP::CS2-CS3 |
| Wu215_R | TGCGTAGCGTTACAGTACCTGAT | |
| Giulia007 | acggccagtgaattcgagctGTGAAGCGGGTCCGGGTG | 53G-SP, CVD 1233-SP and CVD 1233-SP::CS2-CS3 |
| GP173 | ATGGAAACCACCGTATTTCTCAGCAACCGCAGC | |
| GP174 | GCTGCGGTTGCTGAGAAATACGGTGGTTTCCAT | |
| JM296 | AACTTCAGCATCAGAATGACTCCCTTTC | |
| JM298 | GTCATTCTGATGCTGAAGTTTATGCTCGATACCAAC | |
| GM174 | TGTGTAGGCTGGAGCTGCTT | |
| GM175 | ATGGGAATTAGCCATGGTCC | |
| Giulia010 | ccatggctaattcccatTCAGCTCCAGTCTTCAGTTC | |
| Giulia011 | agcctacacaCCTGTTCATCAGAAATCATCTC | |
| GP141 | CATGATTACGCCAAGCTgtaatcgtcgtgtgtgg | |
| GP175 | gcaatggcctgtgtcacc | |
| GP176 | atgtggctgtccattgcc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilla, G.; Wu, T.; Grassel, C.; Moon, J.; Foulke-Abel, J.; Tang, C.M.; Barry, E.M. Evaluation of a Live Attenuated S. sonnei Vaccine Strain in the Human Enteroid Model. Pathogens 2021, 10, 1079. https://doi.org/10.3390/pathogens10091079
Pilla G, Wu T, Grassel C, Moon J, Foulke-Abel J, Tang CM, Barry EM. Evaluation of a Live Attenuated S. sonnei Vaccine Strain in the Human Enteroid Model. Pathogens. 2021; 10(9):1079. https://doi.org/10.3390/pathogens10091079
Chicago/Turabian StylePilla, Giulia, Tao Wu, Christen Grassel, Jonathan Moon, Jennifer Foulke-Abel, Christoph M. Tang, and Eileen M. Barry. 2021. "Evaluation of a Live Attenuated S. sonnei Vaccine Strain in the Human Enteroid Model" Pathogens 10, no. 9: 1079. https://doi.org/10.3390/pathogens10091079
APA StylePilla, G., Wu, T., Grassel, C., Moon, J., Foulke-Abel, J., Tang, C. M., & Barry, E. M. (2021). Evaluation of a Live Attenuated S. sonnei Vaccine Strain in the Human Enteroid Model. Pathogens, 10(9), 1079. https://doi.org/10.3390/pathogens10091079