
MicroRNA-155 Modulates Macrophages’ Response to Non-Tuberculous Mycobacteria through COX-2/PGE2 Signaling




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
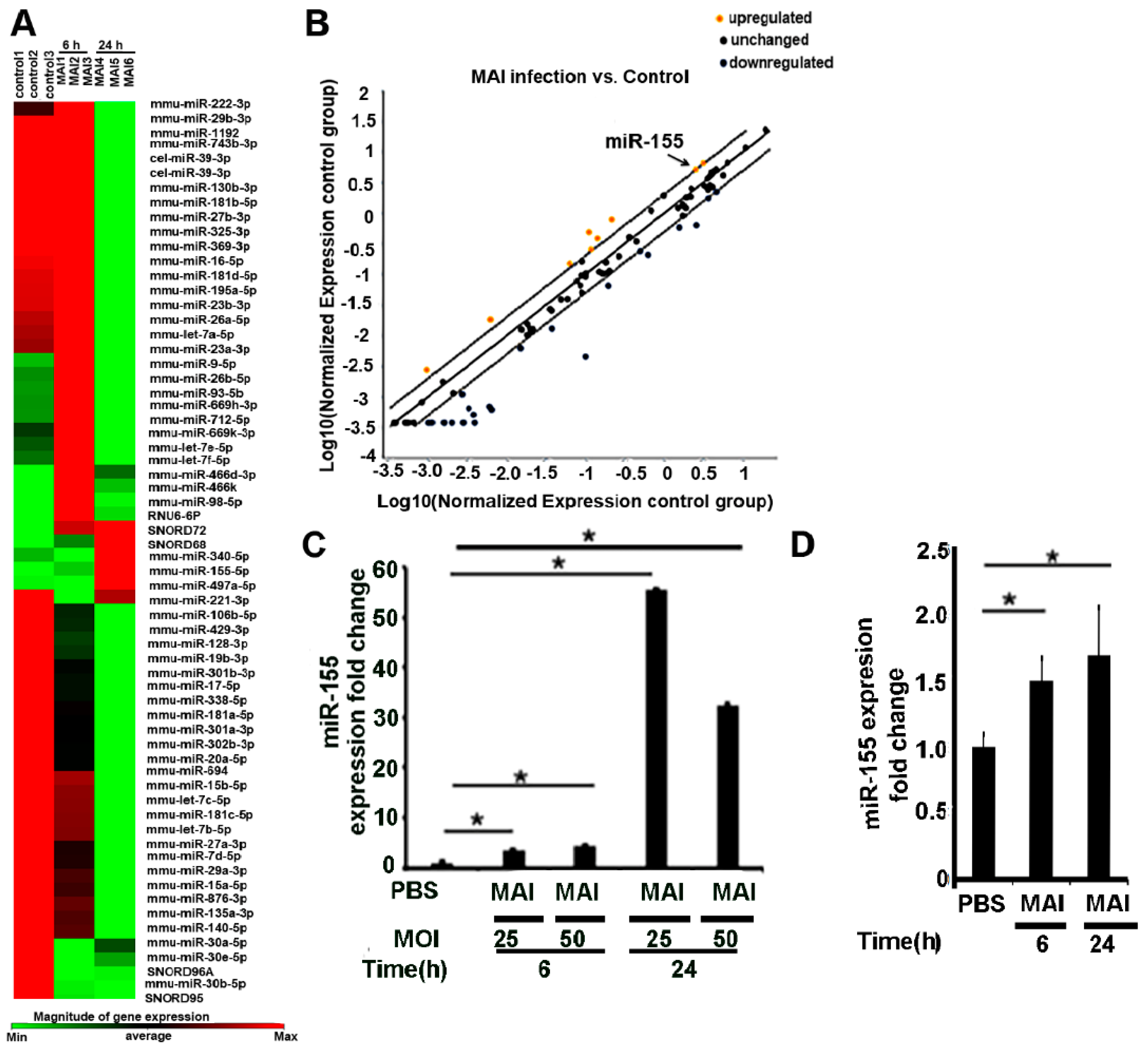
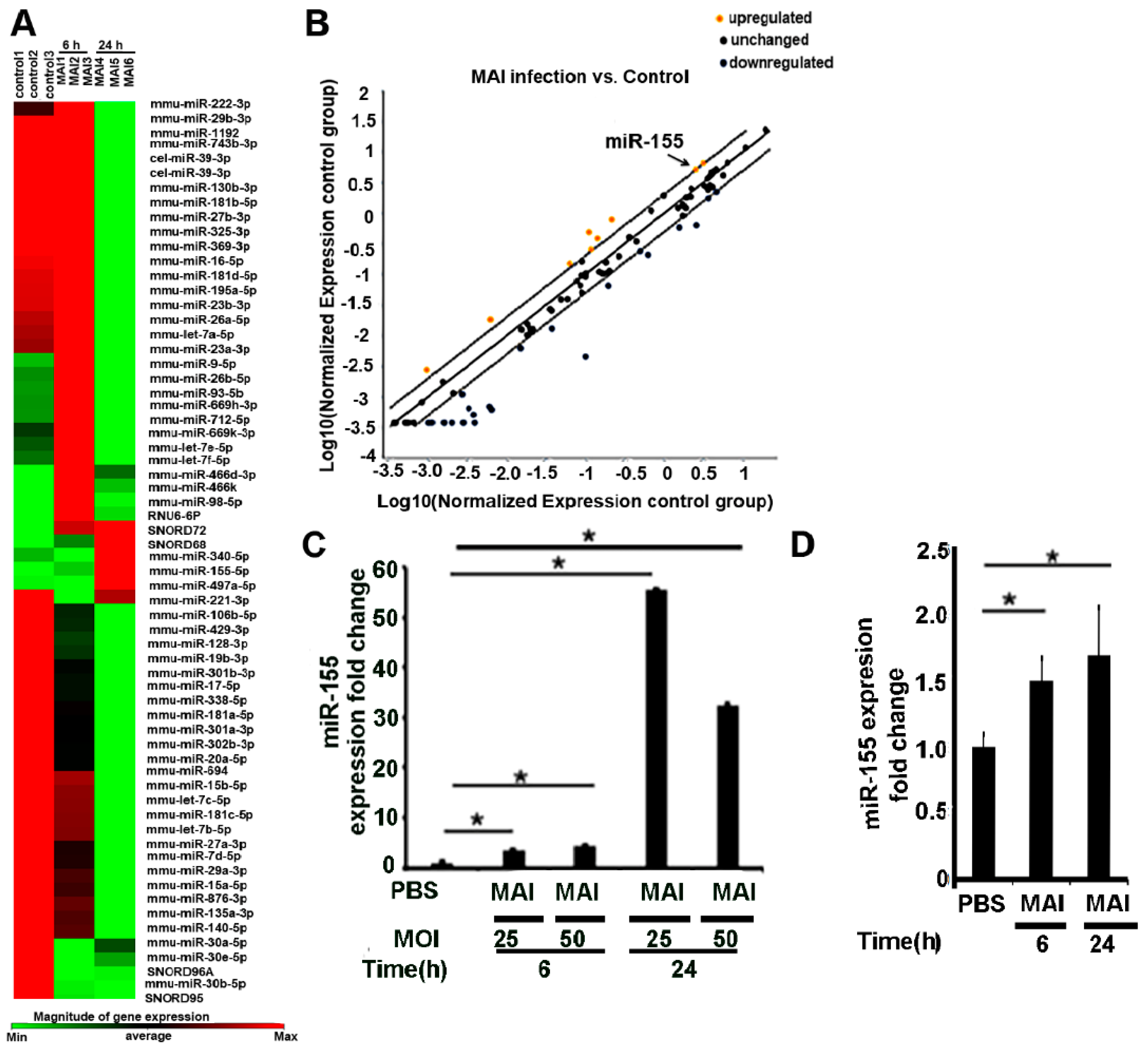
2.1. MiR-155 Is Upregulated in Bone-Marrow-Derived Macrophages (BMDM) Infected with MAI
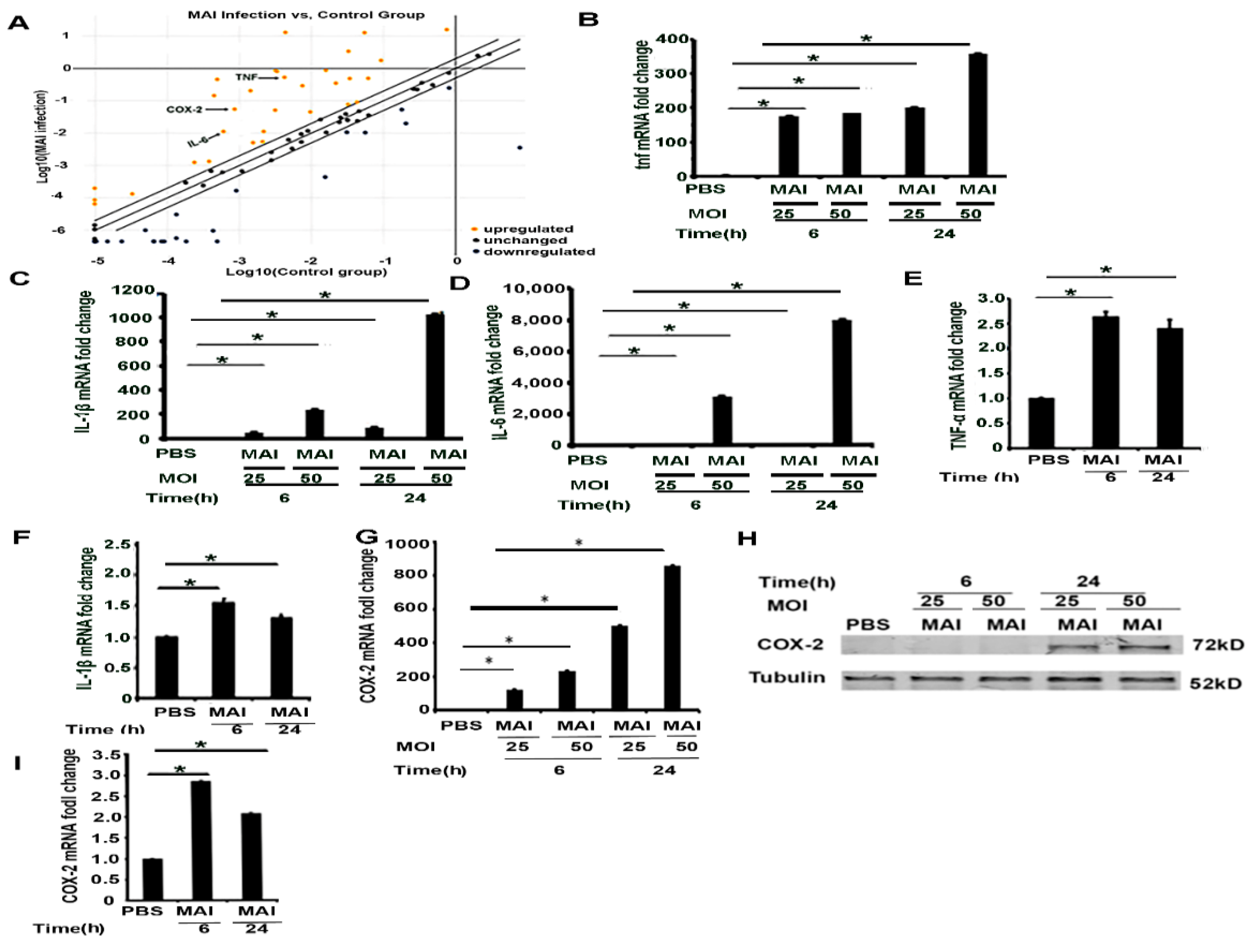
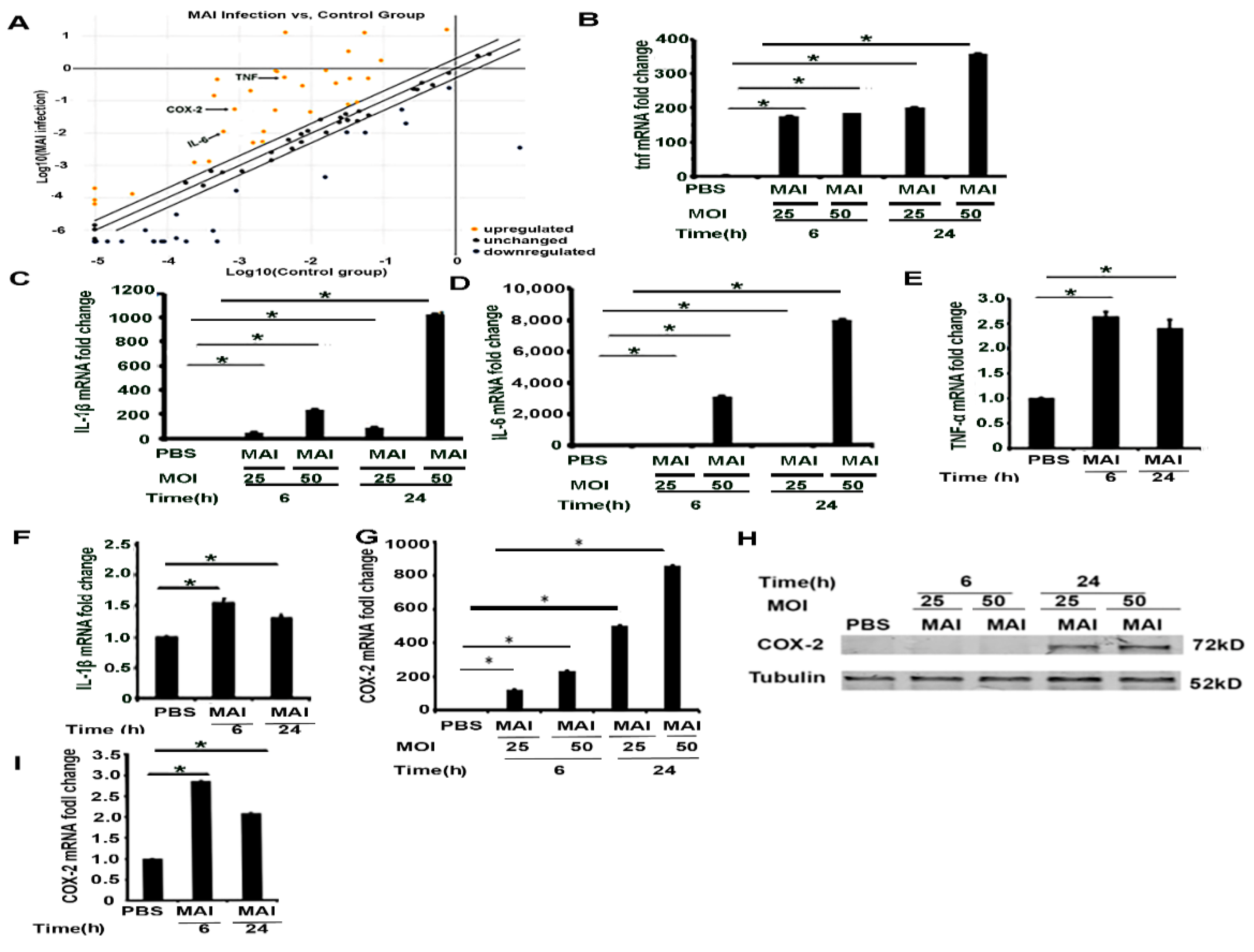
2.2. Pro-Inflammatory Mediators Regulated by miR-155 Are Induced in Macrophages Infected with MAI
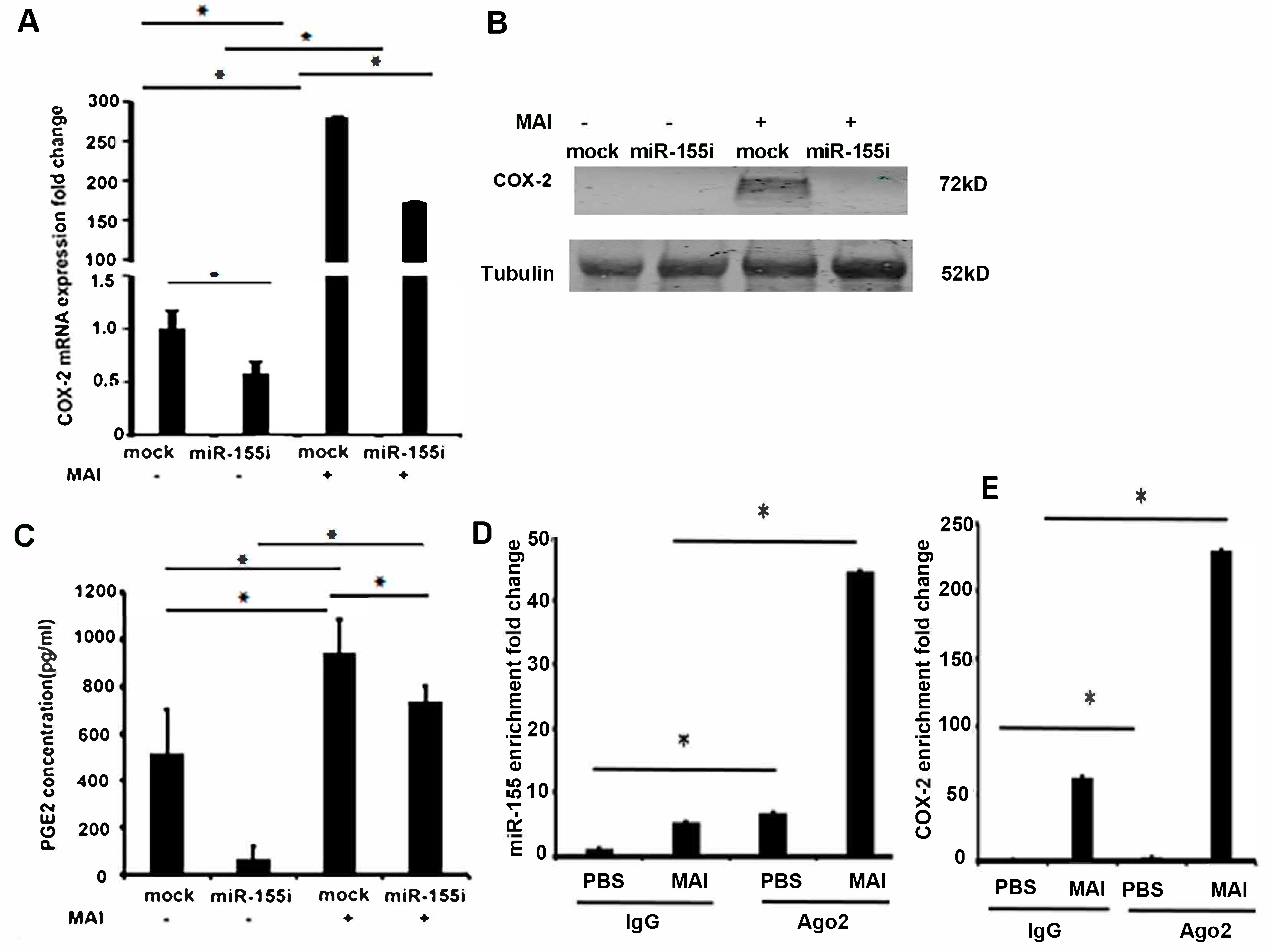
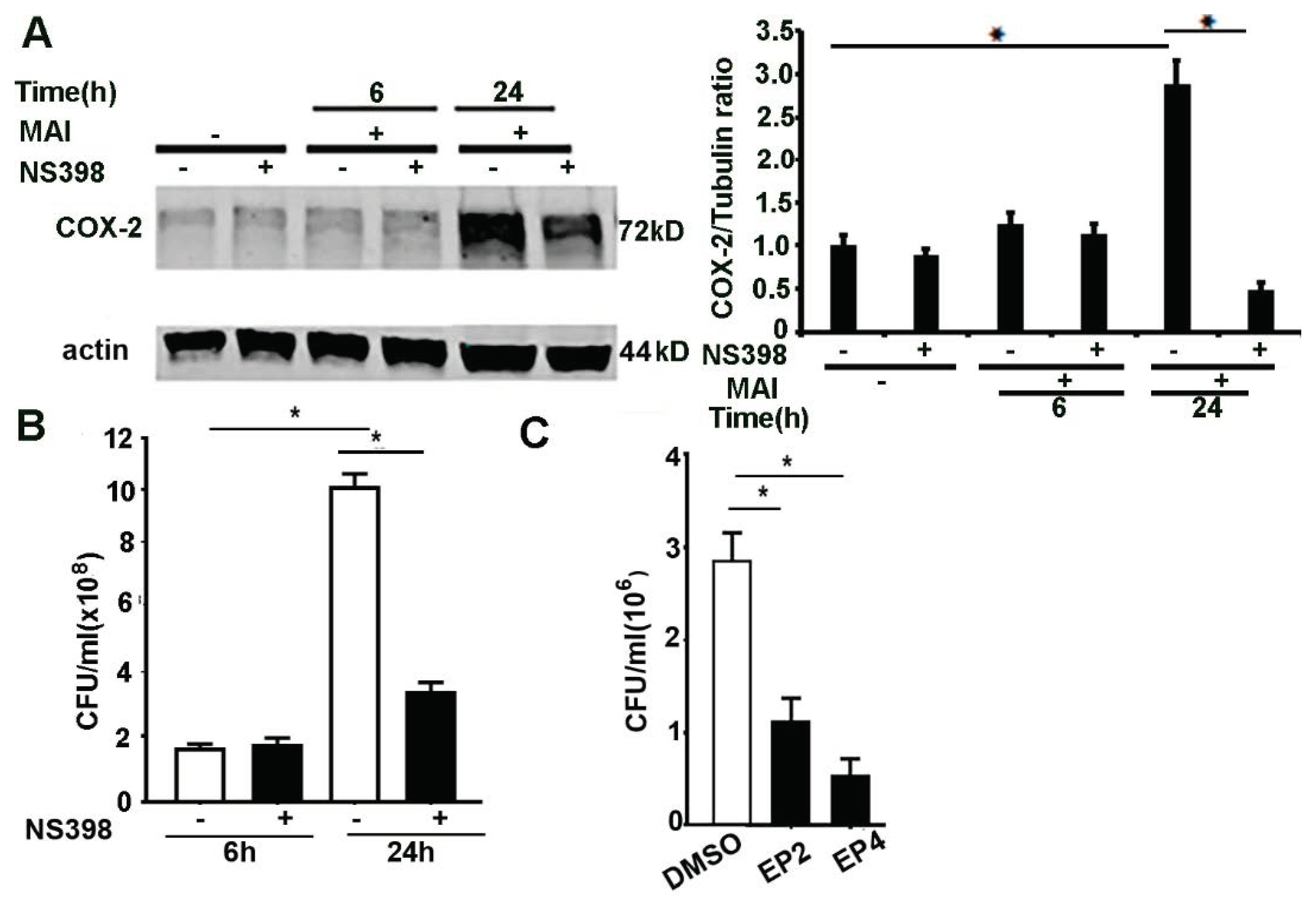
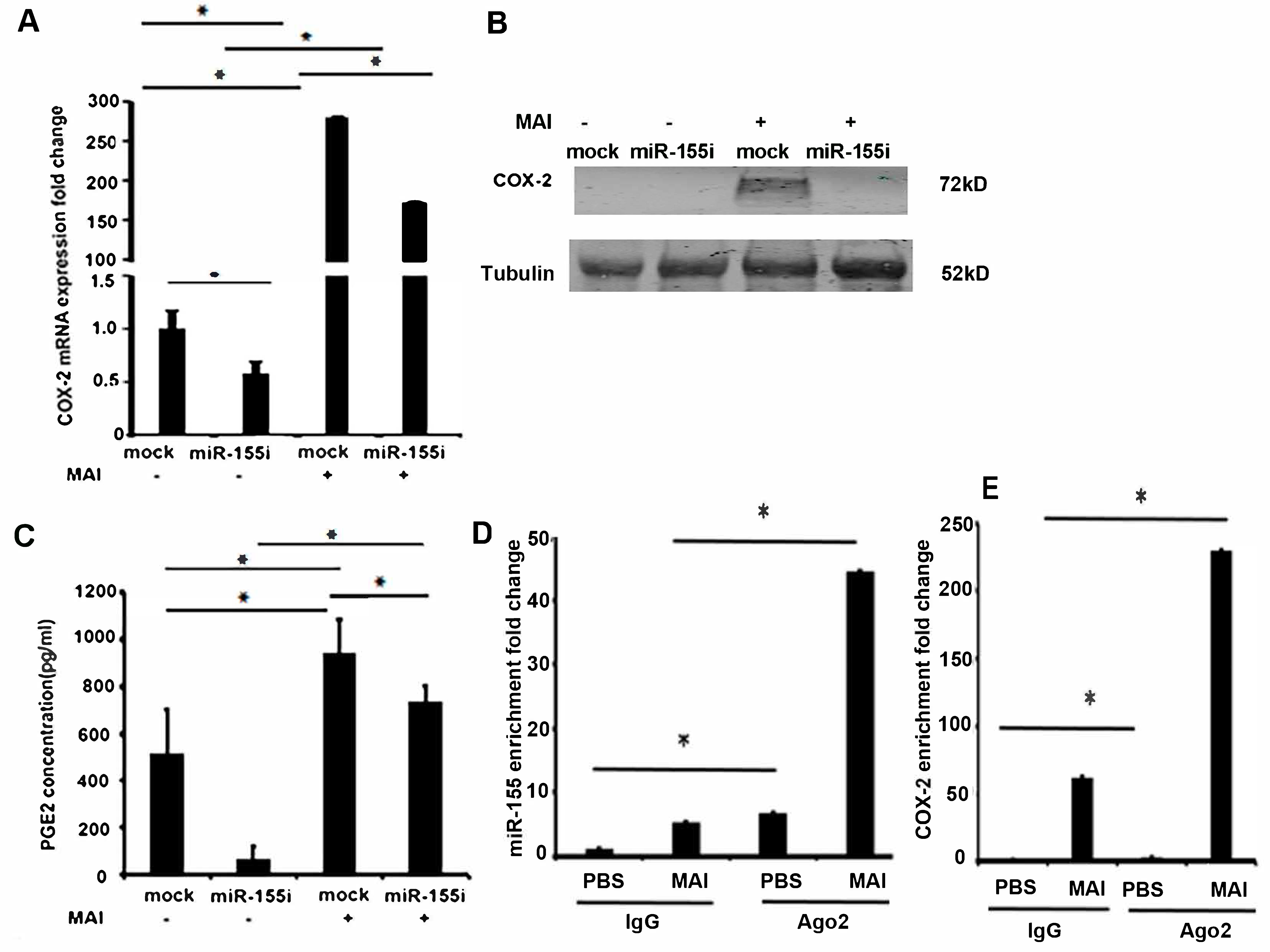
2.3. Induction of COX-2 and Production of PGE2 by MAI Is Dependent on miR-155
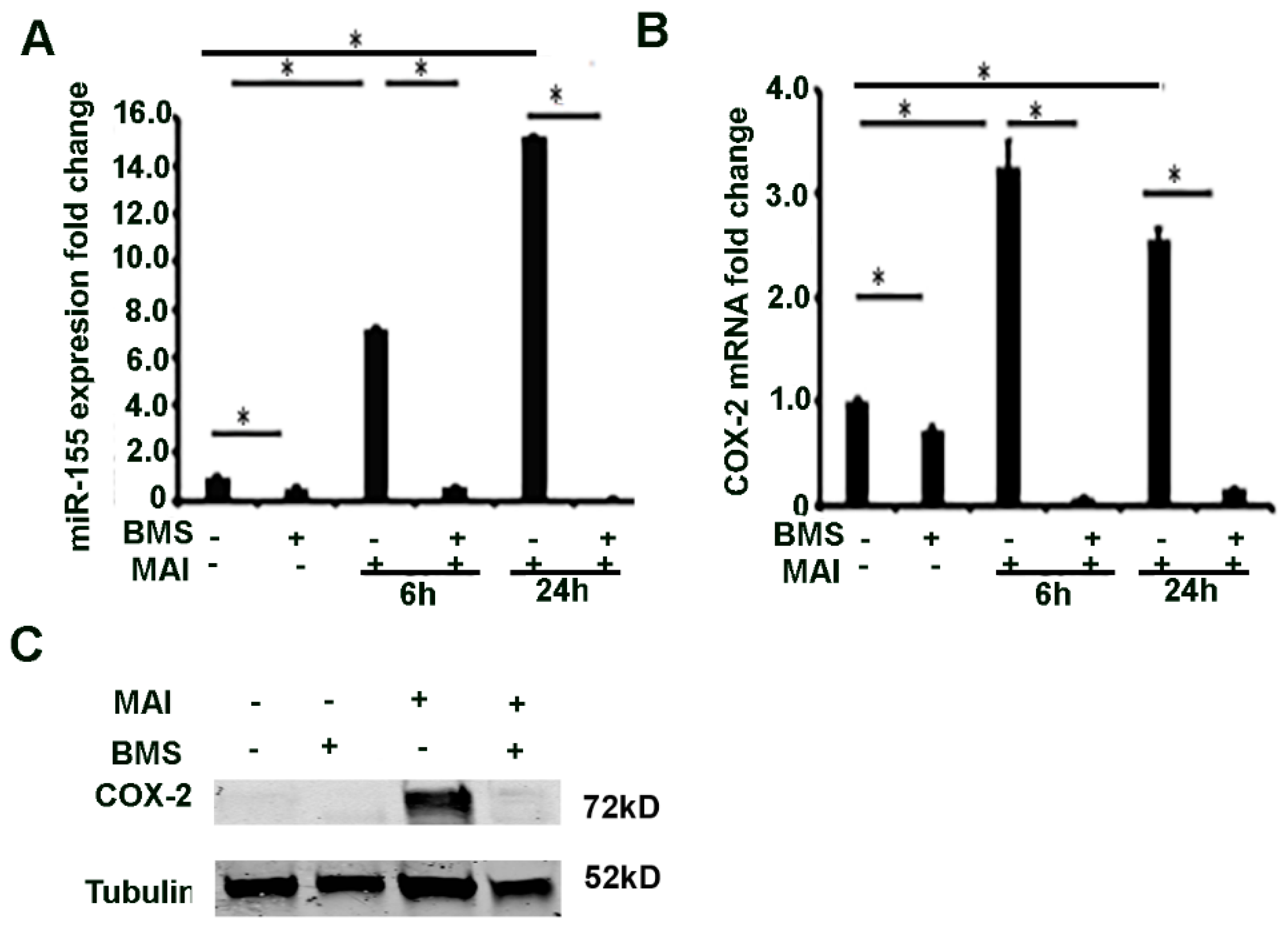
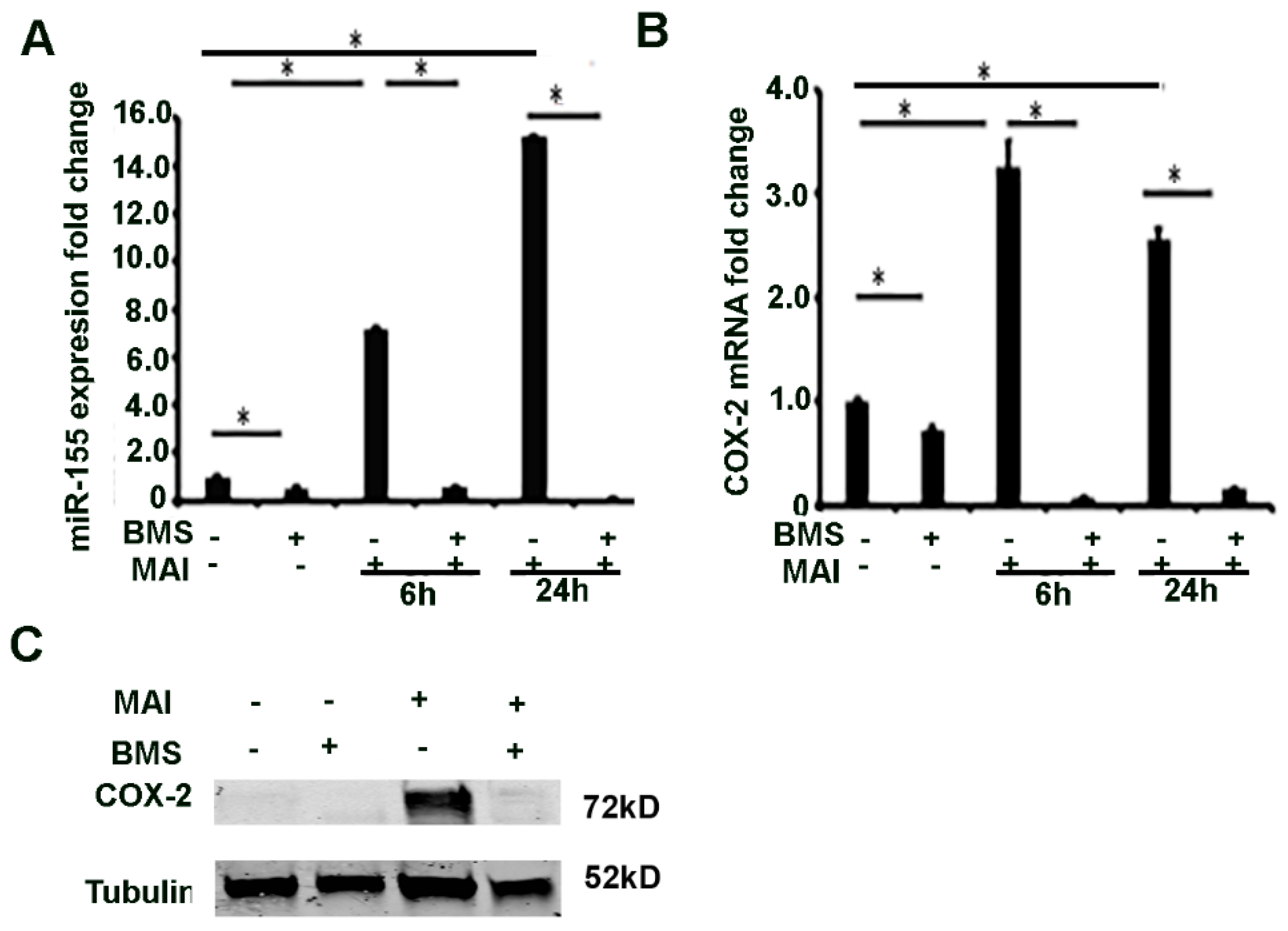
2.4. MiR-155 and COX-2 Expression Are Transcriptionally Regulated by NF-kB in Macrophages Infected with MAI
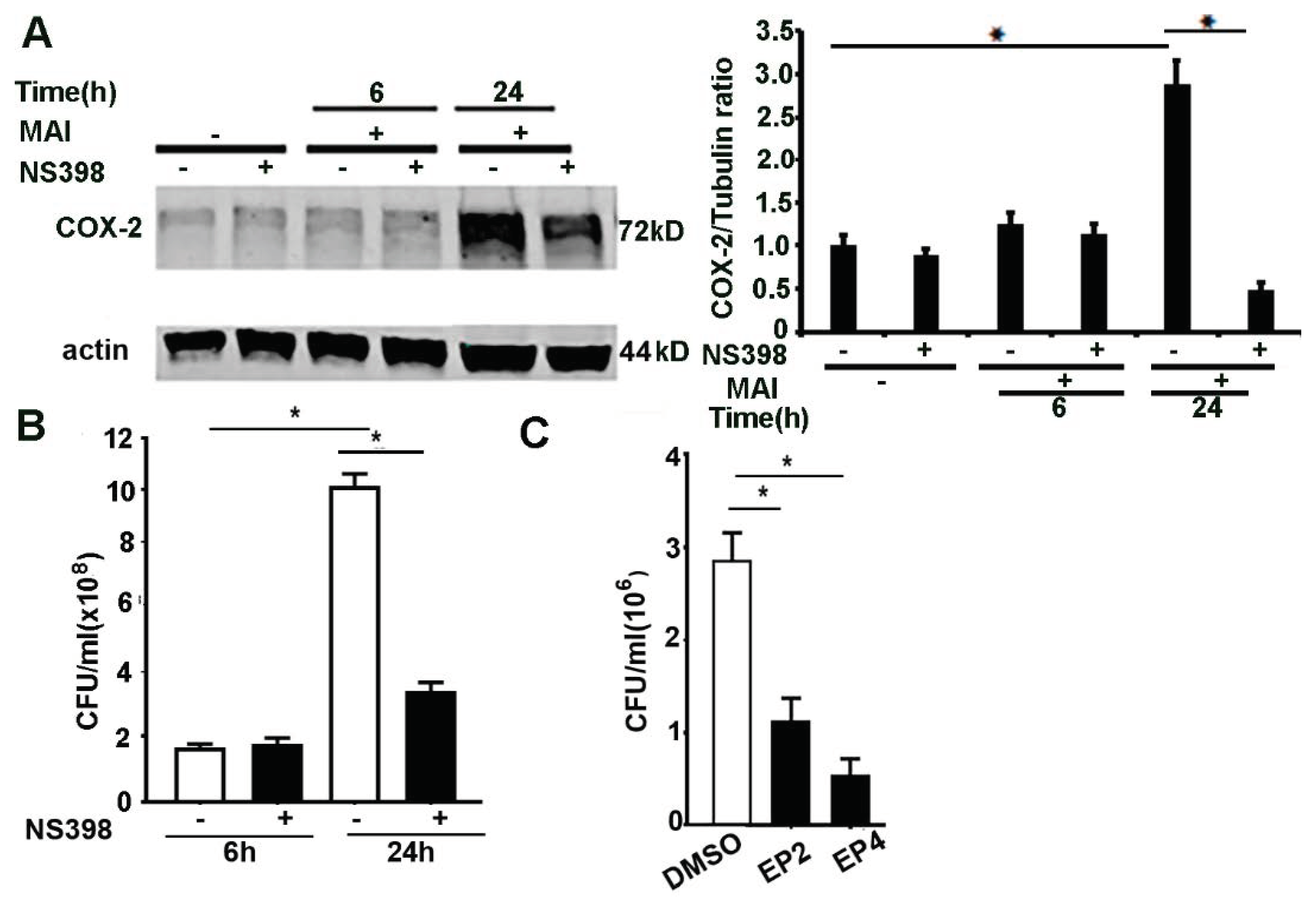
2.5. Inhibition of COX-2, EP2 and EP4 Enhances Killing of MAI in Macrophages
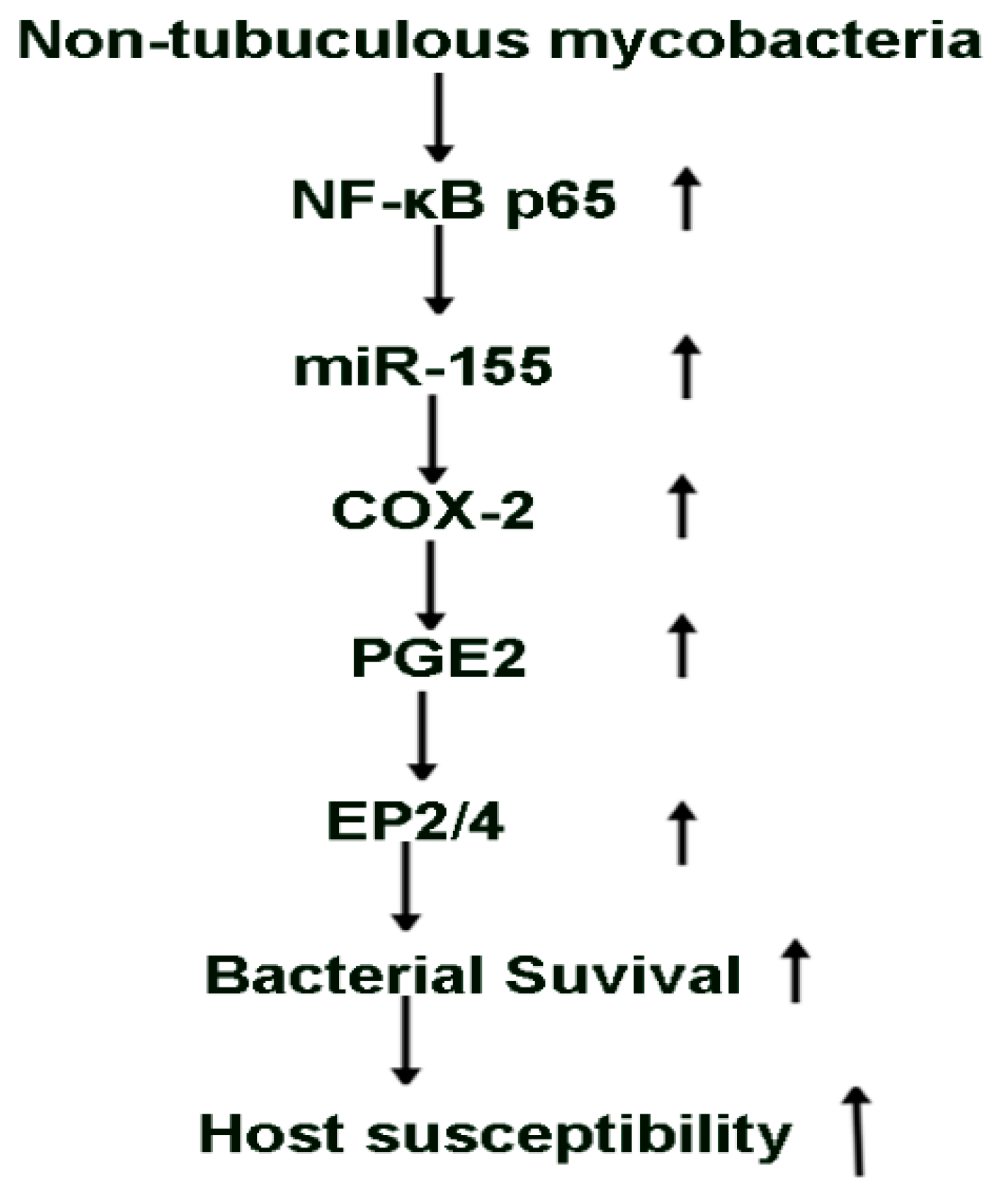
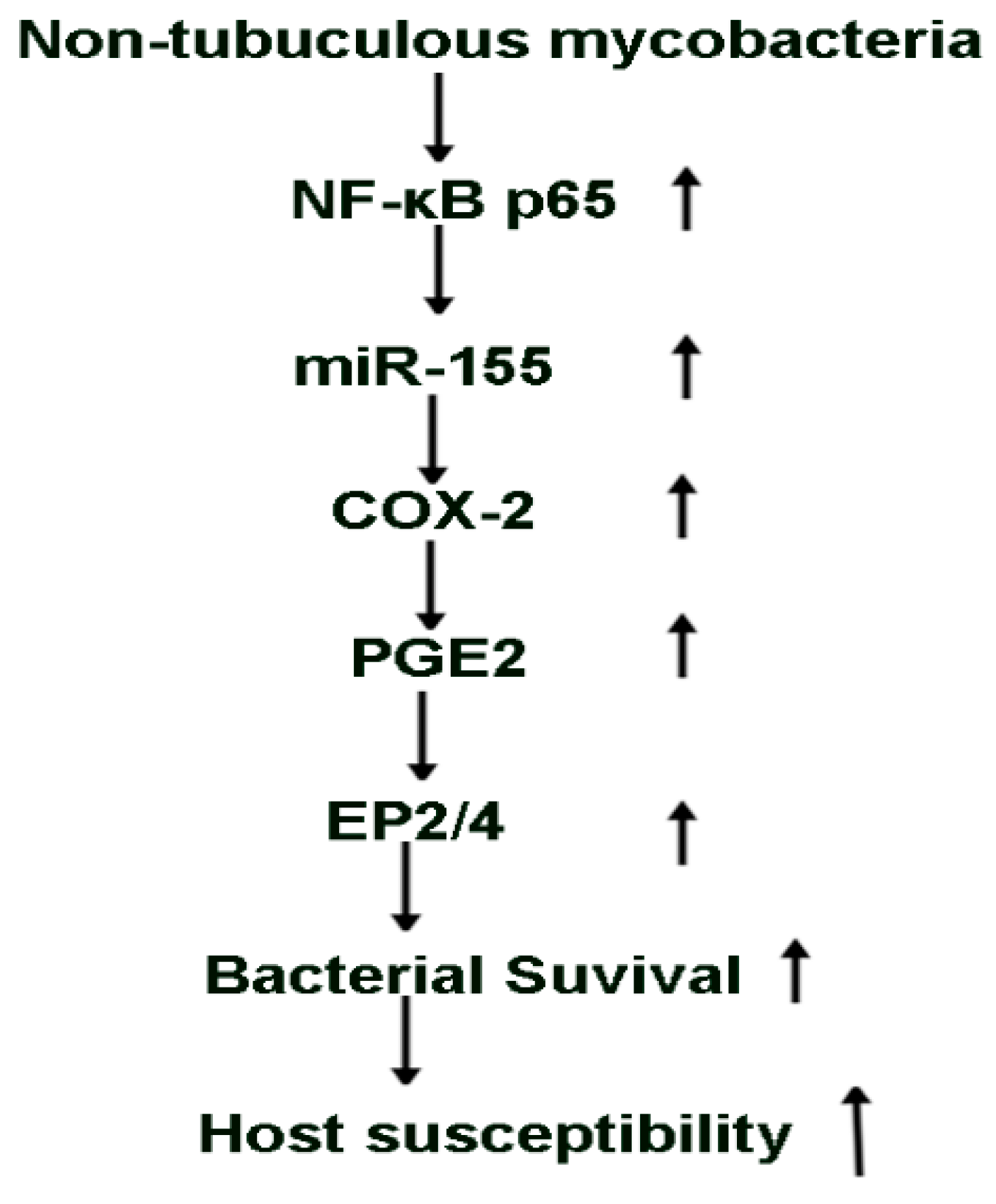
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bacterial Strain
4.3. Ethics Statement
4.4. Animal
4.5. Cell Culture and Treatment
4.6. MiRNAs Primer-Profiling PCR Array
4.7. RT2 Profiler™ PCR Array
4.8. Quantitative RT-PCR
4.9. Western Blot Analysis
4.10. ELISA
4.11. RNA Binding Protein Immunoprecipitation (RIP) Assay
4.12. Bacterial Survival Assay
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Floto, R.A.; Olivier, K.N.; Saiman, L.; Daley, C.L.; Herrmann, J.L.; Nick, J.A.; Noone, P.G.; Bilton, D.; Corris, P.; Gibson, R.L.; et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis: Executive summary. Thorax 2016, 71, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Char, A.; Hopkinson, N.S.; Hansell, D.M.; Nicholson, A.G.; Shaw, E.C.; Clark, S.J.; Sedgwick, P.; Wilson, R.; Jordan, S.; Loebinger, M.R. Evidence of mycobacterial disease in COPD patients with lung volume reduction surgery; the importance of histological assessment of specimens: A cohort study. BMC Pulm. Med. 2014, 14, 124. [Google Scholar] [CrossRef] [Green Version]
- Marras, T.K.; Campitelli, M.A.; Kwong, J.C.; Lu, H.; Brode, S.K.; Marchand-Austin, A.; Gershon, A.S.; Jamieson, F.B. Risk of Nontuberculous Mycobacterial Pulmonary Disease with Obstructive Lung Disease. Eur. Respir. J. 2016, 48, 928–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel-Wayman, S.; Abate, G.; Barber, D.L.; Bermudez, L.E.; Coler, R.N.; Cynamon, M.H.; Daley, C.L.; Davidson, R.M.; Dick, T.; Floto, R.A.; et al. Advancing Translational Science for Pulmonary Nontuberculous Mycobacterial Infections. A Road Map for Research. Am. J. Respir. Crit. Care Med. 2019, 199, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, D.S.; Leyland, R.; Kurowska-Stolarska, M.; Patil, S.A.; Balaji, K.N. MicroRNA-155 Is Required for Mycobacterium bovis BCG-Mediated Apoptosis of Macrophages. Mol. Cell. Biol. 2012, 32, 2239–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, C.L.; Iaccarino, J.M.; Lange, C.; Cambau, E.; Jr, R.J.W.; Andrejak, C.; Böttger, E.C.; Brozek, J.; Griffith, D.E.; Guglielmetti, L.; et al. Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official ATS/ERS/ESCMID/IDSA Clinical Practice Guideline. Eur. Respir. J. 2020, 56, 2000535. [Google Scholar] [CrossRef] [PubMed]
- Van Ingen, J.; Wagner, D.; Gallagher, J.; Morimoto, K.; Lange, C.; Haworth, C.S.; Floto, R.A.; Adjemian, J.; Prevots, D.R.; Griffith, D.E. Poor Adherence to Management Guidelines in Nontuberculous Mycobacterial Pulmonary Diseases. Eur. Respir. J. 2016, 49, 1601855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirsaeidi, M.; Hadid, W.; Ericsoussi, B.; Rodgers, D.; Sadikot, R.T. Non-Tuberculous Mycobacterial Disease Is Common in Patients With Non-Cystic Fibrosis Bronchiectasis. Int. J. Infect. Dis. 2013, 17, e1000–e1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasla, Z.; Sutliff, R.L.; Sadikot, R.T. Macrophage Signaling Pathways in Pulmonary Nontuberculous Mycobacteria Infections. Am. J. Respir. Cell Mol. Biol. 2020, 63, 144–151. [Google Scholar] [CrossRef]
- Flume, P.A.; Griffith, D.E.; Chalmers, J.D.; Daley, C.L.; Olivier, K.; O’Donnell, A.; Aksamit, T.; Kasperbauer, S.; Leitman, A.; Winthrop, K.L. Development of Drugs for Nontuberculous Mycobacterial Disease. Chest 2021, 159, 537–543. [Google Scholar] [CrossRef]
- Aronoff, D.M.; Lewis, C.; Serezani, C.H.; Eaton, K.A.; Goel, D.; Phipps, J.C.; Peters-Golden, M.; Mancuso, P. E-Prostanoid 3 Receptor Deletion Improves Pulmonary Host Defense and Protects Mice from Death in Severe Streptococcus pneumoniae Infection. J. Immunol. 2009, 183, 2642–2649. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Colón, G.J.; Moore, B.B. Prostaglandin E 2 As a Regulator of Immunity to Pathogens. Pharmacol. Ther. 2018, 185, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.A.T.; Trindade, B.C.; Secatto, A.; Nicolete, R.; Peres-Buzalaf, C.; Ramos, S.G.; Sadikot, R.; Bitencourt, C.D.S.; Faccioli, L.H. Celecoxib Improves Host Defense through Prostaglandin Inhibition during Histoplasma capsulatum Infection. Mediat. Inflamm. 2013, 2013, 950981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Panchal, D.; Syed, M.A.; Mehta, H.; Joo, M.; Hadid, W.; Sadikot, R.T. Induction of Cyclooxygenase-2 Signaling by Stomatococcus mucilaginosus Highlights the Pathogenic Potential of an Oral Commensal. J. Immunol. 2013, 191, 3810–3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadikot, R.T.; Zeng, H.; Azim, A.C.; Joo, M.; Dey, S.K.; Breyer, R.M.; Peebles, R.S.; Blackwell, T.S.; Christman, J.W. Bacterial Clearance of Pseudomonas aeruginosa Is Enhanced by the Inhibition of COX-2. Eur. J. Immunol. 2007, 37, 1001–1009. [Google Scholar] [CrossRef]
- Joo, M.; Kwon, M.; Sadikot, R.T.; Kingsley, P.J.; Marnett, L.J.; Blackwell, T.S.; Peebles, R.S.; Urade, Y.; Christman, J.W. Induction and Function of Lipocalin Prostaglandin D Synthase in Host Immunity. J. Immunol. 2007, 179, 2565–2575. [Google Scholar] [CrossRef]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.; Remold, H.G. Lipid Mediators in Innate Immunity against Tuberculosis: Opposing Roles of PGE2 and LXA4 in the Induction of Macrophage Death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xiong, X.; Zhai, W.; Zhu, T.; Zhu, X.; Zhu, Y.; Peng, Y.; Zhang, Y.; Wang, J.; Chen, H.; et al. Upregulation of Cytokines and Differentiation of Th17 and Treg by Dendritic Cells: Central Role of Prostaglandin E2 Induced by Mycobacterium bovis. Microorganisms 2020, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Divangahi, M.; Desjardins, D.; Nunes-Alves, C.; Remold, H.G.; Behar, S.M. Eicosanoid Pathways Regulate Adaptive Immunity to Mycobacterium tuberculosis. Nat. Immunol. 2010, 11, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nat. Cell Biol. 2014, 511, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zuo, M.; Chen, H.; Liu, S.; Wu, X.; Cui, Z.; Yang, H.; Liu, H.; Ge, B. Mycobacterium tuberculosis Lipoprotein MPT83 Induces Apoptosis of Infected Macrophages by Activating the TLR2/p38/COX-2 Signaling Pathway. J. Immunol. 2017, 198, 4772–4780. [Google Scholar] [CrossRef] [Green Version]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Assis, P.A.; Espíndola, M.S.; Paula-Silva, F.W.; Rios, W.M.; Pereira, P.A.; Leão, S.C.; Silva, C.L.; Faccioli, L.H. Mycobacterium tuberculosis expressing phospholipase C subverts PGE2 synthesis and induces necrosis in alveolar macrophages. BMC Microbiol. 2014, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Wen, Q.; Du, X.; Wang, J.; He, W.; Wang, R.; Hu, S.; Zhou, X.; Yang, J.; Gao, Y.; et al. Novel Function of Cyclooxygenase-2: Suppressing Mycobacteria by Promoting Autophagy via the Protein Kinase B/Mammalian Target of Rapamycin Pathway. J. Infect. Dis. 2018, 217, 1267–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, M.; Sadikot, R.T. PGD synthase and PGD2 in immune resposne. Med. Inflamm. 2012, 2012, 503128. [Google Scholar] [CrossRef] [Green Version]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorhoi, A.; Iannaccone, M.; Farinacci, M.; Faé, K.C.; Schreiber, J.; Moura-Alves, P.; Nouailles, G.; Mollenkopf, H.J.; Oberbeck-Müller, D.; Jörg, S.; et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J. Clin. Investig. 2013, 123, 4836–4848. [Google Scholar] [CrossRef] [Green Version]
- Singh, Y.; Kaul, V.; Mehra, A.; Chatterjee, S.; Tousif, S.; Dwivedi, V.P.; Suar, M.; Van Kaer, L.; Bishai, W.R.; Das, G. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J. Biol. Chem. 2013, 288, 5056–5061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Song, Z.; Wu, Y.; Gao, Y.; Gao, M.; Liu, F.; Wang, F.; Zhang, Y. MicroRNA-27b Modulates Inflammatory Response and Apoptosis during Mycobacterium tuberculosis Infection. J. Immunol. 2018, 200, 3506–3518. [Google Scholar] [CrossRef] [Green Version]
- Alipoor, S.D.; Adcock, I.M.; Tabarsi, P.; Folkerts, G.; Mortaz, E. MiRNAs in tuberculosis: Their decisive role in the fate of TB. Eur. J. Pharmacol. 2020, 886, 173529. [Google Scholar] [CrossRef]
- Ruiz-Tagle, C.; Naves, R.; Balcells, M.E. Unraveling the Role of MicroRNAs in Mycobacterium tuberculosis Infection and Disease: Advances and Pitfalls. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Huang, C.; Xu, X.; Teng, J. miR-155 Knockdown Protects against Cerebral Ischemia and Reperfusion Injury by Targeting MafB. BioMed Res. Int. 2020, 2020, 6458204. [Google Scholar] [CrossRef]
- Vigorito, E.; Kohlhaas, S.; Lu, D.; Leyland, R. miR-155: An ancient regulator of the immune system. Immunol. Rev. 2013, 253, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Syed, M.; Panchal, D.; Joo, M.; Bedi, C.; Lim, S.; Onyuksel, H.; Rubinstein, I.; Colonna, M.; Sadikot, R.T. TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am. J. Physiol. Lung Cell. Molecul. Physiol. 2016, 310, L426–L438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, S.R.; Mangan, N.E.; Caffrey, B.E.; Gantier, M.P.; Williams, B.R.; Hertzog, P.J.; McCoy, C.E.; O’Neill, L.A. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J. Biol. Chem. 2014, 289, 4316–4325. [Google Scholar] [CrossRef] [Green Version]
- Rothchild, A.C.; Sissons, J.R.; Shafiani, S.; Plaisier, C.; Min, D.; Mai, D.; Gilchrist, M.; Peschon, J.; Larson, R.P.; Bergthaler, A.; et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, E6172–E6181. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Zhang, Y.; Do, D.C.; Ke, X.; Zhang, S.; Lambert, K.; Kumar, S.; Hu, C.; Zhou, Y.; Ishmael, F.T.; et al. miR-155 Modulates Cockroach Allergen- and Oxidative Stress-Induced Cyclooxygenase-2 in Asthma. J. Immunol. 2018, 201, 916–929. [Google Scholar] [CrossRef]
- Comer, B.S. Does miRNA-155 Promote Cyclooxygenase-2 Expression in Cancer? Drug Dev. Res. 2015, 76, 354–356. [Google Scholar] [CrossRef] [Green Version]
- Comer, B.S.; Camoretti-Mercado, B.; Kogut, P.C.; Halayko, A.J.; Solway, J.; Gerthoffer, W.T. Cyclooxygenase-2 and microRNA-155 expression are elevated in asthmatic airway smooth muscle cells. Am. J. Respir. Cell Molecul. Biol. 2015, 52, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, T.; Newstead, M.W.; Zeng, X.; Nanjo, Y.; Peters-Golden, M.; Kaku, M.; Standiford, T.J. Interleukin-36γ and IL-36 receptor signaling mediate impaired host immunity and lung injury in cytotoxic Pseudomonas aeruginosa pulmonary infection: Role of prostaglandin E2. PLoS Pathog. 2017, 13, e1006737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, S.; Qi, H.H.; Chowdhury, D.; Shi, Y.; Novina, C.D. Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins. Nat. Struct. Mol. Biol. 2009, 16, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.M. Structure and function of argonaute proteins. Structure 2005, 13, 1403–1408. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Molecul. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Serezani, C.H.; Chung, J.; Ballinger, M.N.; Moore, B.B.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 2007, 37, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Aronoff, D.M.; Bergin, I.L.; Lewis, C.; Goel, D.; O’Brien, E.; Peters-Golden, M.; Mancuso, P. E-prostanoid 2 receptor signaling suppresses lung innate immunity against Streptococcus pneumoniae. Prostaglandins Other Lipid mediat. 2012, 98, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Nore, K.G.; Jørgensen, M.J.; Dyrhol-Riise, A.M.; Jenum, S.; Tonby, K. Elevated Levels of Anti-Inflammatory Eicosanoids and Monocyte Heterogeneity in Mycobacterium tuberculosis Infection and Disease. Front. Immun. 2020, 11, 579849. [Google Scholar] [CrossRef]
- Hata, A.N.; Breyer, R.M. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004, 103, 147–166. [Google Scholar] [CrossRef]
- Aronoff, D.M.; Canetti, C.; Peters-Golden, M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immun. 2004, 173, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Mehta, H.J.; Mohammed, K.; Nasreen, N.; Roman, R.; Brantly, M.; Sadikot, R.T. TREM-1 is induced in tumor associated macrophages by cyclo-oxygenase pathway in human non-small cell lung cancer. PLoS ONE 2014, 9, e94241. [Google Scholar] [CrossRef]
- Naqvi, R.A.; Gupta, M.; George, A.; Naqvi, A.R. MicroRNAs in shaping the resolution phase of inflammation. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, K.; Jia, Q.; Zhang, H.; Bie, Q.; Zhang, B. The Roles of Host Noncoding RNAs in Mycobacterium tuberculosis Infection. Front. Immun. 2021, 12, 664787. [Google Scholar] [CrossRef]
- Shamaei, M.; Mirsaeidi, M. Nontuberculous mycobacteria, macrophages, and host innate immune response. Infect. Immun. 2021, IAI-00812. [Google Scholar] [CrossRef]
- Zhang, C.; Xi, X.; Wang, Q.; Jiao, J.; Zhang, L.; Zhao, H.; Lai, Z. The association between serum miR-155 and natural killer cells from tuberculosis patients. Int. J. Clin. Exp. Med. 2015, 8, 9168–9172. [Google Scholar]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, K.; Zhou, L.; Wu, Y.; Zhu, M.; Lai, X.; Chen, T.; Feng, L.; Li, M.; Huang, C.; et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 2013, 9, e1003697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, J.; Moore, S.A.; Ballas, Z.K.; Portanova, J.P.; Krieg, A.M.; Berg, D.J. CpG DNA induces cyclooxygenase-2 expression and prostaglandin production. Int. Immunol. 2001, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Reis, C.S. Distinct roles of eicosanoids in the immune response to viral encephalitis: Or why you should take NSAIDS. Viral immunol. 2002, 15, 133–146. [Google Scholar] [CrossRef]
- Bowman, C.C.; Bost, K.L. Cyclooxygenase-2-mediated prostaglandin E2 production in mesenteric lymph nodes and in cultured macrophages and dendritic cells after infection with Salmonella. J. Immunol. 2004, 172, 2469–2475. [Google Scholar] [CrossRef]
- Fredenburgh, L.E.; Velandia, M.M.; Ma, J.; Olszak, T.; Cernadas, M.; Englert, J.A.; Chung, S.W.; Liu, X.; Begay, C.; Padera, R.F.; et al. Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis. J. Immunol. 2011, 187, 5255–5267. [Google Scholar] [CrossRef] [PubMed]
- Brogliato, A.R.; Antunes, C.A.; Carvalho, R.S.; Monteiro, A.P.; Tinoco, R.F.; Bozza, M.T.; Canetti, C.; Peters-Golden, M.; Kunkel, S.L.; Vianna-Jorge, R.; et al. Ketoprofen impairs immunosuppression induced by severe sepsis and reveals an important role for prostaglandin E2. Shock 2012, 38, 620–629. [Google Scholar] [CrossRef]
- Nakajima, M.; Matsuyama, M.; Kawaguchi, M.; Kiwamoto, T.; Matsuno, Y.; Morishima, Y.; Yoshida, K.; Sherpa, M.; Yazaki, K.; Osawa, H.; et al. Nrf2 Regulates Granuloma Formation and Macrophage Activation during Mycobacterium avium Infection via Mediating Nramp1 and HO-1 Expressions. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Syed, M.A.; Panchal, D.; Joo, M.; Colonna, M.; Brantly, M.; Sadikot, R.T. Triggering receptor expressed on myeloid cells 1 (TREM-1)-mediated Bcl-2 induction prolongs macrophage survival. J. Biol. Chem. 2014, 289, 15118–15129. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Wang, J.J.; Qi, M.; Yoon, J.J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Fan, X.; Staitieh, B.; Bedi, C.; Spearman, P.; Guidot, D.M.; Sadikot, R.T. HIV-related proteins prolong macrophage survival through induction of Triggering receptor expressed on myeloid cells-1. Sci. Rep. 2017, 7, 42028. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Prasla, Z.; Lee, F.E.-H.; Bedi, B.; Sutliff, R.L.; Sadikot, R.T. MicroRNA-155 Modulates Macrophages’ Response to Non-Tuberculous Mycobacteria through COX-2/PGE2 Signaling. Pathogens 2021, 10, 920. https://doi.org/10.3390/pathogens10080920
Yuan Z, Prasla Z, Lee FE-H, Bedi B, Sutliff RL, Sadikot RT. MicroRNA-155 Modulates Macrophages’ Response to Non-Tuberculous Mycobacteria through COX-2/PGE2 Signaling. Pathogens. 2021; 10(8):920. https://doi.org/10.3390/pathogens10080920
Chicago/Turabian StyleYuan, Zhihong, Zohra Prasla, Frances Eun-Hyung Lee, Brahmchetna Bedi, Roy L. Sutliff, and Ruxana T. Sadikot. 2021. "MicroRNA-155 Modulates Macrophages’ Response to Non-Tuberculous Mycobacteria through COX-2/PGE2 Signaling" Pathogens 10, no. 8: 920. https://doi.org/10.3390/pathogens10080920
APA StyleYuan, Z., Prasla, Z., Lee, F. E.-H., Bedi, B., Sutliff, R. L., & Sadikot, R. T. (2021). MicroRNA-155 Modulates Macrophages’ Response to Non-Tuberculous Mycobacteria through COX-2/PGE2 Signaling. Pathogens, 10(8), 920. https://doi.org/10.3390/pathogens10080920