
Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria

 , and
, and
Abstract
1. Introduction
2. Results
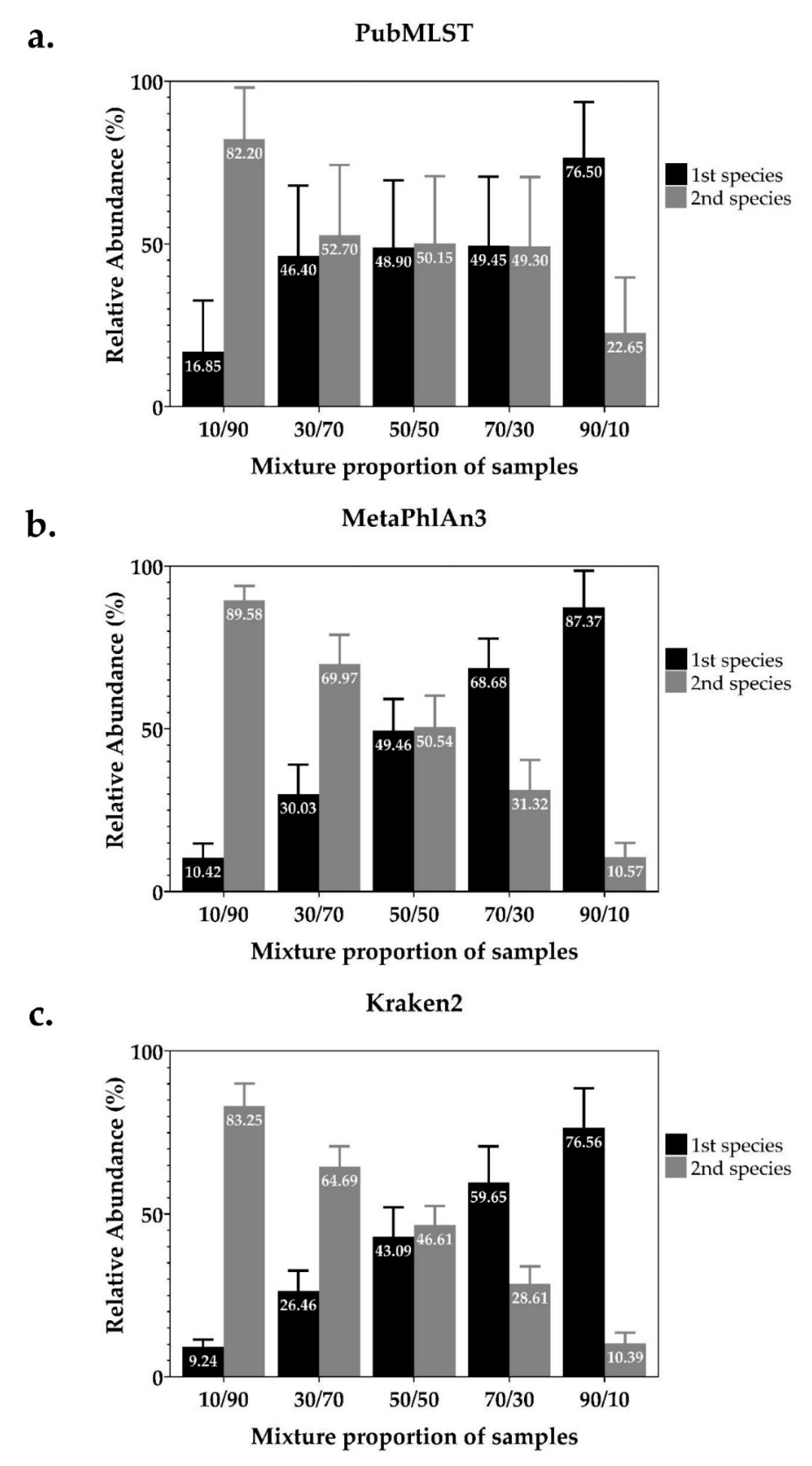
2.1. Detection and Identification of Mixed Infections from In-Silico Simulated Mixed-Species NTM Dataset
2.2. Detection and Identification of NTM Species in Mixed Infections: Dataset from GenBank
2.3. Comparison of the Four WGS-Analysis Tools for Identifying Mixed Infections with Different NTM Species
2.4. Concordance between WGS-Analysis Tools and LPA for Detection and Identification of Mixed NTM Species in Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Study Population: Clinical Samples of Mixed NTM Species
4.2. Sample Preparation and WGS of Clinical Samples
4.3. In-Silico Simulated Samples Containing Various Proportions of Reads from Different NTM Species
4.4. WGS Data Samples from a Public Database
4.5. Bioinformatics Analysis
4.5.1. QC Check and Data Preparation of Sequence Reads
4.5.2. Detection of Mixed Species of NTM Using Analysis of Read Frequencies Supporting SNP Alleles
4.5.3. Detection of Mixed Species of NTM Using PubMLST
4.5.4. Detection of Mixed Species of NTM Using MetaPhlAn3
4.5.5. Detection of Mixed Species of NTM Using Kraken2
4.5.6. Detection of Mixed Species of NTM Using Mykrobe-Predictor
4.5.7. Detection of Mixed Species of NTM Using Metagenomic Assembly Analysis Based on 16S rRNA, rpoB and hsp65 Genes
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wagner, D.; Young, L. Nontuberculous mycobacterial infections: A clinical review. Infection 2004, 32, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Inamdar, L.; O’riordain, D.; Schweiger, M.; Watson, J. Nontuberculous mycobacteria in non-HIV patients: Epidemiology, treatment and response. Eur. Respir. J. 2004, 23, 741–746. [Google Scholar] [CrossRef]
- Hirabayashi, R.; Nakagawa, A.; Takegawa, H.; Tomii, K. A case of pleural effusion caused by Mycobacterium fortuitum and Mycobacterium mageritense coinfection. BMC Infect. Dis. 2019, 19, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Jhun, B.W.; Kim, S.-Y.; Choe, J.; Jeon, K.; Huh, H.J.; Ki, C.-S.; Lee, N.Y.; Shin, S.J.; Daley, C.L. Nontuberculous mycobacterial lung diseases caused by mixed infection with Mycobacterium avium complex and Mycobacterium abscessus complex. Antimicrob. Agents Chemother. 2018, 62, e01105-18. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-J.; Park, C.M.; Park, Y.S.; Lee, J.; Lee, S.-M.; Yang, S.-C.; Yoo, C.-G.; Kim, Y.W.; Han, S.K.; Yim, J.-J. Isolation of multiple nontuberculous mycobacteria species in the same patients. Int. J. Infect. Dis. 2011, 15, e795–e798. [Google Scholar] [CrossRef]
- Wallace, R.J., Jr.; Zhang, Y.; Brown, B.A.; Dawson, D.; Murphy, D.T.; Wilson, R.; Griffith, D.E. Polyclonal Mycobacterium avium complex infections in patients with nodular bronchiectasis. Am. J. Respir. Crit. Care Med. 1998, 158, 1235–1244. [Google Scholar] [CrossRef]
- Prevots, D.R.; Marras, T.K. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: A review. Clin. Chest Med. 2015, 36, 13–34. [Google Scholar] [CrossRef]
- Singh, A.K.; Marak, R.S.; Maurya, A.K.; Das, M.; Nag, V.L.; Dhole, T.N. Mixed cutaneous infection caused by Mycobacterium szulgai and Mycobacterium intermedium in a healthy adult female: A rare case report. Case Rep. Dermatol. Med. 2015, 2015, 607519. [Google Scholar] [CrossRef][Green Version]
- Bekou, V.; Büchau, A.; Flaig, M.J.; Ruzicka, T.; Hogardt, M. Cutaneous infection by Mycobacterium haemophilum and kansasii in an IgA-deficient man. BMC Dermatol. 2011, 11, 1–5. [Google Scholar] [CrossRef]
- Lévy-Frébault, V.; Pangon, B.; Buré, A.; Katlama, C.; Marche, C.; David, H. Mycobacterium simiae and Mycobacterium avium-M. intracellulare mixed infection in acquired immune deficiency syndrome. J. Clin. Microbiol. 1987, 25, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Häfner, B.; Haag, H.; Geiss, H.-K.; Nolte, O. Different molecular methods for the identification of rarely isolated non-tuberculous mycobacteria and description of new hsp65 restriction fragment length polymorphism patterns. Mol. Cell. Probes 2004, 18, 59–65. [Google Scholar] [CrossRef]
- Kirschner, P.; Bottger, E.C. Species identification of mycobacteria using rDNA sequencing. In Mycobacteria Protocols; Springer: Berlin/Heidelberg, Germany, 1998; pp. 349–361. [Google Scholar]
- Kim, B.-J.; Lee, S.-H.; Lyu, M.-A.; Kim, S.-J.; Bai, G.-H.; Kim, S.-J.; Chae, G.-T.; Kim, E.-C.; Cha, C.-Y.; Kook, Y.-H. Identification of mycobacterial species by comparative sequence analysis of the RNA polymerase gene (rpoB). J. Clin. Microbiol. 1999, 37, 1714. [Google Scholar] [CrossRef] [PubMed]
- Ringuet, H.; Akoua-Koffi, C.; Honore, S.; Varnerot, A.; Vincent, V.; Berche, P.; Gaillard, J.; Pierre-Audigier, C. hsp65 sequencing for identification of rapidly growing mycobacteria. J. Clin. Microbiol. 1999, 37, 852. [Google Scholar] [CrossRef]
- Dai, J.; Chen, Y.; Dean, S.; Morris, J.G.; Salfinger, M.; Johnson, J.A. Multiple-genome comparison reveals new loci for Mycobacterium species identification. J. Clin. Microbiol. 2011, 49, 144. [Google Scholar] [CrossRef]
- Hwang, S.M.; Lim, M.S.; Hong, Y.J.; Kim, T.S.; Park, K.U.; Song, J.; Lee, J.H.; Kim, E.C. Simultaneous detection of Mycobacterium tuberculosis complex and nontuberculous mycobacteria in respiratory specimens. Tuberculosis 2013, 93, 642–646. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, B.; Du, C.; Tian, X.; Cai, X.; Hou, Y.; Li, H.; Zheng, R.; Li, J.; Liu, Y. Rapid identification of clinically relevant Mycobacterium species by multicolor melting curve analysis. J. Clin. Microbiol. 2019, 57, e01096-18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shin, J.H. Identification of Nontuberculous Mycobacteria from Clinical Isolates and Specimens using AdvanSure Mycobacteria GenoBlot Assay. Jpn. J. Infect. Dis. 2020, 73, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Gan, M.; Liu, Q.; Yang, C.; Gao, Q.; Luo, T. Deep whole-genome sequencing to detect mixed infection of Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0159029. [Google Scholar] [CrossRef]
- Sobkowiak, B.; Glynn, J.R.; Houben, R.M.; Mallard, K.; Phelan, J.E.; Guerra-Assunção, J.A.; Banda, L.; Mzembe, T.; Viveiros, M.; McNerney, R. Identifying mixed Mycobacterium tuberculosis infections from whole genome sequence data. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Quan, T.P.; Bawa, Z.; Foster, D.; Walker, T.; Del Ojo Elias, C.; Rathod, P.; Iqbal, Z.; Bradley, P.; Mowbray, J.; Walker, A.S. Evaluation of whole-genome sequencing for mycobacterial species identification and drug susceptibility testing in a clinical setting: A large-scale prospective assessment of performance against line probe assays and phenotyping. J. Clin. Microbiol. 2018, 56, e01480-17. [Google Scholar] [CrossRef]
- Pfeiffer, W.; Braun, J.; Burchell, J.; Witte, C.L.; Rideout, B.A. Whole-genome analysis of mycobacteria from birds at the San Diego Zoo. PLoS ONE 2017, 12, e0173464. [Google Scholar] [CrossRef]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef]
- Pankhurst, L.J.; Del Ojo Elias, C.; Votintseva, A.A.; Walker, T.M.; Cole, K.; Davies, J.; Fermont, J.M.; Gascoyne-Binzi, D.M.; Kohl, T.A.; Kong, C. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: A prospective study. Lancet Respir. Med. 2016, 4, 49–58. [Google Scholar] [CrossRef]
- Fedrizzi, T.; Meehan, C.J.; Grottola, A.; Giacobazzi, E.; Serpini, G.F.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; Bertorelli, R.; De Sanctis, V. Genomic characterization of nontuberculous mycobacteria. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Doster, E.; Rovira, P.; Noyes, N.R.; Burgess, B.A.; Yang, X.; Weinroth, M.D.; Linke, L.; Magnuson, R.; Boucher, C.; Belk, K.E. A cautionary report for pathogen identification using shotgun metagenomics; a comparison to aerobic culture and polymerase chain reaction for Salmonella enterica identification. Front. Microbiol. 2019, 10, 2499. [Google Scholar] [CrossRef] [PubMed]
- Vijayvargiya, P.; Jeraldo, P.R.; Thoendel, M.J.; Greenwood-Quaintance, K.E.; Esquer Garrigos, Z.; Sohail, M.R.; Chia, N.; Pritt, B.S.; Patel, R. Application of metagenomic shotgun sequencing to detect vector-borne pathogens in clinical blood samples. PLoS ONE 2019, 14, e0222915. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-K.; Kim, T.S.; Kim, J.-I.; Yim, J.-J. Whole genome sequencing of Nontuberculous Mycobacterium (NTM) isolates from sputum specimens of co-habiting patients with NTM pulmonary disease and NTM isolates from their environment. BMC Genomics 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinforma. 2019, 20, 1125–1136. [Google Scholar] [CrossRef]
- Miossec, M.J.; Valenzuela, S.L.; Pérez-Losada, M.; Johnson, W.E.; Crandall, K.A.; Castro-Nallar, E. Evaluation of computational methods for human microbiome analysis using simulated data. PeerJ 2020, 8, e9688. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef]
- Haworth, C.S.; Banks, J.; Capstick, T.; Fisher, A.J.; Gorsuch, T.; Laurenson, I.F.; Leitch, A.; Loebinger, M.R.; Milburn, H.J.; Nightingale, M. British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 2017, 72, ii1–ii64. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Thomas, A.M.; Manghi, P.; Valles-Colomer, M. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. bioRxiv 2020, 10, e65088. [Google Scholar]
- Zinkernagel, M.S.; Zysset-Burri, D.C.; Keller, I.; Berger, L.E.; Leichtle, A.B.; Largiadèr, C.R.; Fiedler, G.M.; Wolf, S. Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Rebollar, E.A.; Gutiérrez-Preciado, A.; Noecker, C.; Eng, A.; Hughey, M.C.; Medina, D.; Walke, J.B.; Borenstein, E.; Jensen, R.V.; Belden, L.K. The skin microbiome of the neotropical frog Craugastor fitzingeri: Inferring potential bacterial-host-pathogen interactions from metagenomic data. Front. Microbiol. 2018, 9, 466. [Google Scholar] [CrossRef] [PubMed]
- Benjak, A.; Avanzi, C.; Benito, Y.; Breysse, F.; Chartier, C.; Boschiroli, M.-L.; Fourichon, C.; Michelet, L.; Pin, D.; Flandrois, J.-P. Highly reduced genome of the new species Mycobacterium uberis, the causative agent of nodular thelitis and tuberculoid scrotitis in livestock and a close relative of the leprosy bacilli. Msphere 2018, 3, e00405-18. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Kinjo, T.; Motooka, D.; Nabeya, D.; Jung, N.; Uechi, K.; Horii, T.; Iida, T.; Fujita, J.; Nakamura, S. Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547 genomic profiles. Emerg. Microbes Infect. 2019, 8, 1043–1053. [Google Scholar] [CrossRef]
- Wuzinski, M.; Bak, A.K.; Petkau, A.; Demczuk, W.H.B.; Soualhine, H.; Sharma, M.K. A multilocus sequence typing scheme for Mycobacterium abscessus complex (MAB-multilocus sequence typing) using whole-genome sequencing data. Int. J. Mycobacteriol. 2019, 8, 273. [Google Scholar]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Mijs, W.; De Vreese, K.; Devos, A.; Pottel, H.; Valgaeren, A.; Evans, C.; Norton, J.; Parker, D.; Rigouts, L.; Portaels, F. Evaluation of a commercial line probe assay for identification of Mycobacterium species from liquid and solid culture. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 794–802. [Google Scholar] [CrossRef]
- Sarkola, A.; Mäkinen, J.; Marjamäki, M.; Marttila, H.; Viljanen, M.; Soini, H. Prospective evaluation of the GenoType assay for routine identification of mycobacteria. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 642–645. [Google Scholar] [CrossRef]
- Jung, H.; Ventura, T.; Chung, J.S.; Kim, W.-J.; Nam, B.-H.; Kong, H.J.; Kim, Y.-O.; Jeon, M.-S.; Eyun, S.-I. Twelve quick steps for genome assembly and annotation in the classroom. PLoS Comput. Biol. 2020, 16, e1008325. [Google Scholar] [CrossRef]
- Huang, Y.; Gilna, P.; Li, W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009, 25, 1338–1340. [Google Scholar] [CrossRef]
- Scarparo, C.; Piccoli, P.; Rigon, A.; Ruggiero, G.; Nista, D.; Piersimoni, C. Direct identification of mycobacteria from MB/BacT alert 3D bottles: Comparative evaluation of two commercial probe assays. J. Clin. Microbiol. 2001, 39, 3222–3227. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E.; Mariottini, A.; Mazzarelli, G. Evaluation of INNO-LiPA MYCOBACTERIA v2: Improved reverse hybridization multiple DNA probe assay for mycobacterial identification. J. Clin. Microbiol. 2003, 41, 4418–4420. [Google Scholar] [CrossRef] [PubMed]
- García-Agudo, L.; Jesús, I.; Rodríguez-Iglesias, M.; García-Martos, P. Evaluation of INNO-LiPA mycobacteria v2 assay for identification of rapidly growing mycobacteria. Braz. J. Microbiol. 2011, 42, 1220–1226. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Larsen, M.H.; Biermann, K.; Tandberg, S.; Hsu, T.; Jacobs, W.R., Jr. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 2007, 6, 10A-2. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Team, S.T.D. Available online: http://ncbi.github.io/sra-tools/ (accessed on 2 March 2021).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. In Multiple Sequence Alignment; Springer: Berlin/Heidelberg, Germany, 2021; pp. 241–260. [Google Scholar]


{kind=link}
| Species Identification Types | PubMLST (n = 100) | MetaPhlAn3 (n = 100) | Kraken2 (n = 100) | Mykrobe-Predictor (n = 100) |
|---|---|---|---|---|
| Correct mixed species | 99 (99%) | 95 (95%) | 100 (100%) | 58 (58%) |
| Incorrect mixed species | 0 | 5 (5%) b | 0 | 25 (25%) |
| Single species only | 1 (1%) | 0 | 0 | 17 (17%) |
| Accession Number | WGS-Analysis Tools | Metagenomic Assembly Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| PubMLST | MetaPhlAn3 | Kraken2 | Mykrobe-Predictor | BLAST (rpoB Gene) | ||||
| Species | RA (%) | Species | RA (%) | Species | RA (%) | Species | Species | |
| SRR5043021 [25] | M. hassiacum, M. peregrinum | 49 39 | M. hassiacum, M. peregrinum | 83.67 16.33 | M. hassiacum, M. peregrinum | 73.41 24.29 | M. hassiacum * | M. hassiacum, M. peregrinum |
| Species Identification Types | PubMLST (n = 155) | MetaPhlAn3 (n = 155) | Kraken2 (n = 155) | Mykrobe-Predictor (n = 155) |
|---|---|---|---|---|
| Correct single species | 149 (96.12%) | 128 (82.58%) | 152 (98.06%) | 149(96.12%) |
| Mixed species | 4 (2.58%) | 26 (16.77%) | 3 (1.93%) | 0 |
| Incorrect species | 1 (0.64%) | 0 | 0 | 5 * (3.22%) |
| No result | 1 (0.64%) | 1 (0.64%) | 0 | 1(0.64%) |
| Sample Groups | Number of Samples (Correct Species Identification/Total Samples) | |||
|---|---|---|---|---|
| PubMLST | MetaPhlAn3 | Kraken2 | Mykrobe-Predictor | |
| Simulated mixed species a | 99/100 | 95/100 | 100/100 | 58/100 |
| Mixed species from public database | 1/1 | 1/1 | 1/1 | 0/1 |
| Sensitivity of tool | 99% (100/101) | 95.04% (96/101) | 100% (101/101) | 57.42% (58/101) |
| Simulated single species control | 15/15 | 14/15 | 15/15 | 14/15 |
| Single species control from public database | 149/155 | 128/155 | 152/155 | 149/155 |
| Specificity of tool | 96.47% (164/170) | 83.52% (142/170) | 98.23% (167/170) | 95.88% (163/170) |
| Sample ID | NTM Species (According to LPA) | Date of Collection | Sex | Age | Specimen Types |
|---|---|---|---|---|---|
| MIX80105 | M. abscessus subsp. massiliense, M. abscessus | 10 January 2014 | M | 43 | Sputum |
| MIX80487 | M. intracellulare, M. scrofulaceum | 16 February 2015 | M | 49 | Nasal Cavity (swab) |
| MIX80628 | M. gordonae, M. simiae | 19 February 2014 | M | 34 | Bone marrow |
| MIX80885 | M. avium, M. intracellulare | 12 March 2014 | M | 63 | Sputum |
| MIX81256 | M. kansasii, M. intracellulare | 8 April 2014 | F | 50 | Bronchial wash |
| MIX81523 | M. kansasii, M. malmeonse | 8 August 2016 | F | 56 | Sputum |
| MIX81666 | M. tuberculosis complex, M. scrofulaceum | 8 July 2016 | M | 69 | Sputum |
| MIX82390 | M. intracellulare, M. avium | 15 July 2014 | M | 34 | Bone marrow |
| S12260 | M. fortuitum, M. peregrinum | 22 August 2016 | F | 75 | Blood |
| S80510 | M. gordonae, M. fortuitum | 17 February 2015 | M | 57 | Sputum |
| S81158 | M. kansasii, M. scrofulaceum | 29 April 2013 | M | 28 | Sputum |
| S81463 | M. simiae, M. fortuitum | 29 May 2013 | F | 61 | Sputum |
| S81801 | M. fortuitum, M. peregrinum | 27 July 2016 | F | 47 | Pus |
| S82945 | M. intracellulare, M. abscessus | 2 October 2013 | F | 81 | Sputum |
| S83359 | M. intracellualre, M. fortuitum | 10 October 2014 | F | 33 | Sputum |
| S83411 | M. kansasii, M. avium | 5 November 2013 | M | 75 | Sputum |
| Sample ID | LPA | WGS-Analysis Tools | |||
|---|---|---|---|---|---|
| PubMLST (MAB-MLST) (%RA) | MetaPhlAn3 (%RA) | Kraken2 (%RA) | Mykrobe-Predictor | ||
| MIX80105 | M. abscessus subsp. massiliense, M. abscessus | M. abscessus (M. abscessus subsp. massiliense) | M. abscessus | M. abscessus | M. abscessus |
| MIX80487 | M. intracellulare, M. scrofulaceum | M. malmoense | M. malmoense | M. malmoense | M. parascrofulaceum |
| MIX80628 | M. gordonae, M. simiae | M. sherrisii, M. asiaticum (96, 3) | M. sherrisii, M. asiaticum M. simiae (99.83, 0.16, 0.01) | M. sherrisii | M. sherrisii |
| MIX80885 | M. avium, M. intracellulare | M. asiaticum | M. sp. 1165178.9, M. lepraemurium, M. colombiense (99.91, 0.06, 0.03) | M. sp. 1165178.9 | M. intracellulare |
| MIX81256 | M. kansasii, M. intracellulare | M. intracellulare | M. intracellulare | M. intracellulare | M. intracellulare |
| MIX81523 | M. kansasii, M. malmeonse | M. attenuatum | M. kansasii | M. sp. MK136 a | M. kansasii |
| MIX81666 | M. tuberculosis complex, M. scrofulaceum | M. tuberculosis | M. canettii b, M. sp. E3198 M. sp 852002-50816 SCH5313054-b (96.70, 1.85, 1.45) | M. tuberculosis | M. tuberculosis M. intracellulare |
| MIX82390 | M. intracellulare, M. avium | M. scrofulaceum | M. lepraemurium, M. scrofulaceum (75.94, 24.06) | M. sp. ACS4054, M. scrofulaceum (9.23, 7.13) | M. intracellulare |
| S12260 | M. fortuitum, M. peregrinum | M. fortuitum | M. fortuitum | M. fortuitum | M. fortuitum |
| S80510 | M. gordonae, M. fortuitum | M. abscessus (UD) | M. gordonae, M. abscessus (81.26, 18.74) | M. gordonae | M. gordonae |
| S81158 | M. kansasii, M. scrofulaceum | M. attenuatum | M. kansasii | M. sp. MK136 a | M. kansasii |
| S81463 | M. simiae, M. fortuitum | No result | M. rhodesiae | M. sp. M26 c | M. farcinogenes |
| S81801 | M. fortuitum, M. peregrinum | M. fortuitum | M. fortuitum, M. abscessus (99.99, 0.01) | M. fortuitum | M. fortuitum |
| S82945 | M. intracellulare, M. abscessus | M. abscessus (M. abscessus subsp. massiliense) | M. abscessus | M. abscessus | M. abscessus |
| S83359 | M. intracellulare, M. fortuitum | M. fortuitum | M. fortuitum | M. fortuitum | M. fortuitum |
| S83411 | M. kansasii, M. avium | M. persicum | M. persicum | M. persicum | M. kansasii |
| Single speciesd | 0 | 14 (87.5%) | 10 (62.5%) | 15 (93.75%) | 15 (93.75%) |
| Mixed speciese | 16(100%) | 1 (6.25%) | 6 (37.5%) | 1 (6.25%) | 1 (6.25%) |
| No result | 0 | 1 (6.25%) | 0 | 0 | 0 |
| Total | 16 | 16 | 16 | 16 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khieu, V.; Ananta, P.; Kaewprasert, O.; Laohaviroj, M.; Namwat, W.; Faksri, K. Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria. Pathogens 2021, 10, 879. https://doi.org/10.3390/pathogens10070879
Khieu V, Ananta P, Kaewprasert O, Laohaviroj M, Namwat W, Faksri K. Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria. Pathogens. 2021; 10(7):879. https://doi.org/10.3390/pathogens10070879
Chicago/Turabian StyleKhieu, Visal, Pimjai Ananta, Orawee Kaewprasert, Marut Laohaviroj, Wises Namwat, and Kiatichai Faksri. 2021. "Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria" Pathogens 10, no. 7: 879. https://doi.org/10.3390/pathogens10070879
APA StyleKhieu, V., Ananta, P., Kaewprasert, O., Laohaviroj, M., Namwat, W., & Faksri, K. (2021). Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria. Pathogens, 10(7), 879. https://doi.org/10.3390/pathogens10070879