
Whole-Genome Sequencing and Comparative Genomics of Three Helicobacter pylori Strains Isolated from the Stomach of a Patient with Adenocarcinoma

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
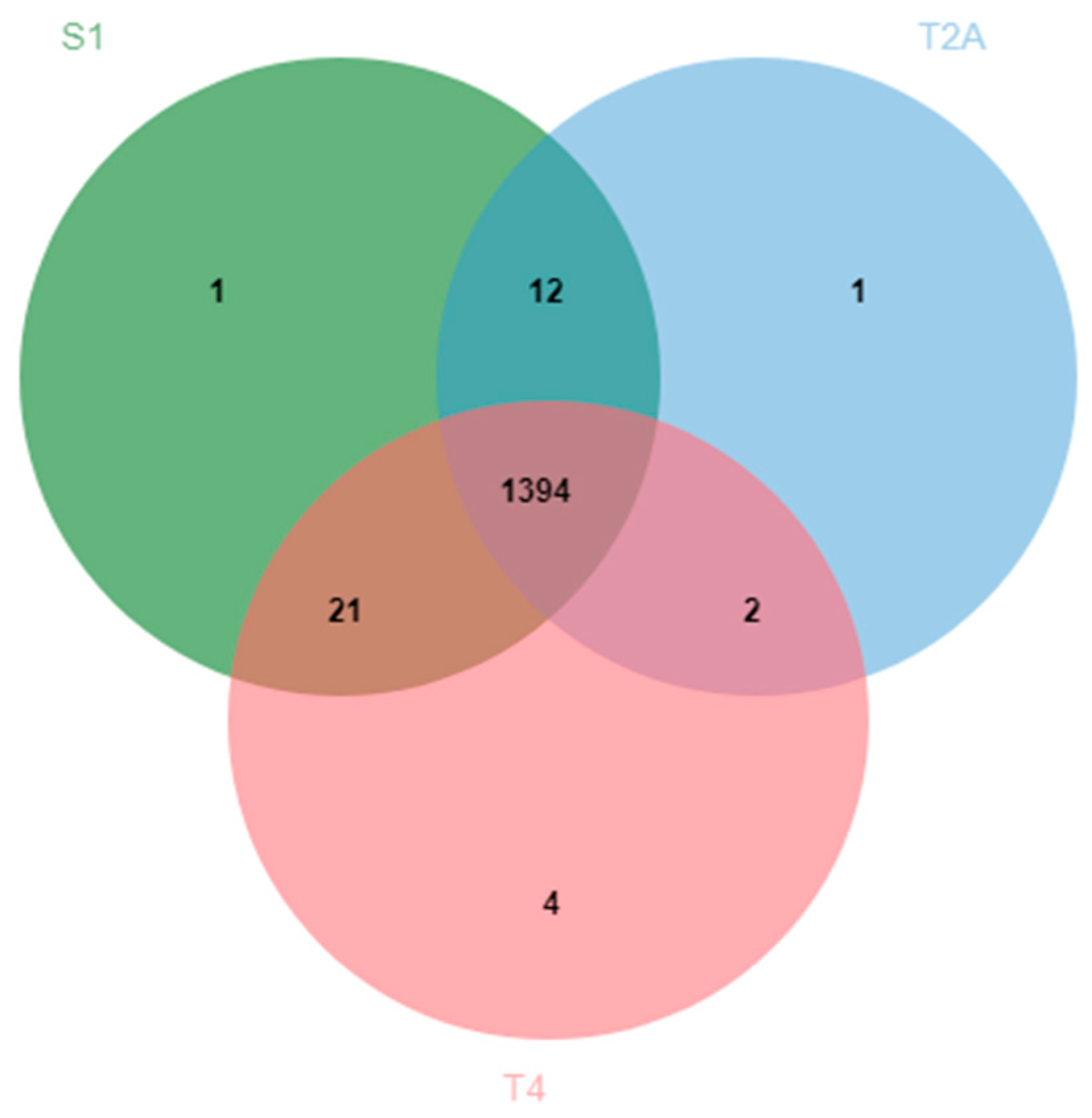
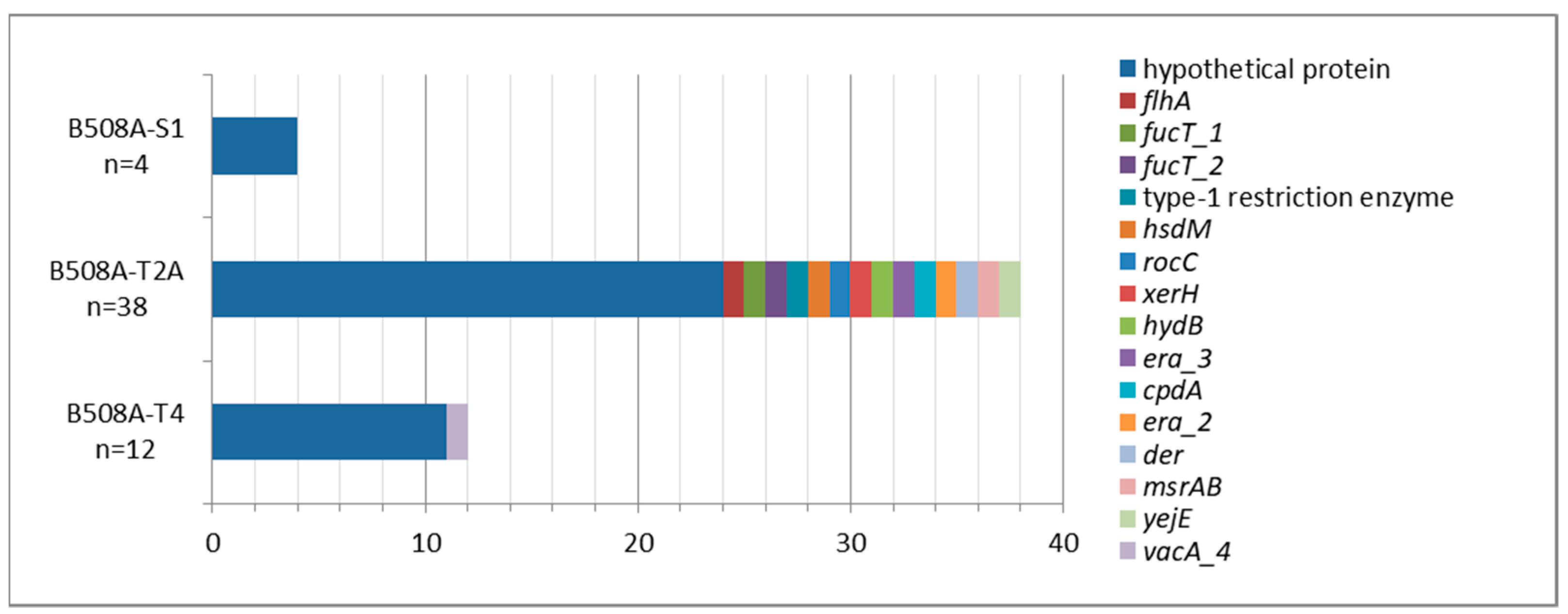
2.1. B508A-S1, B508A-T2A, and B508A-T4 Genome Comparison
2.2. Virulence Factors and Antimicrobial Resistances
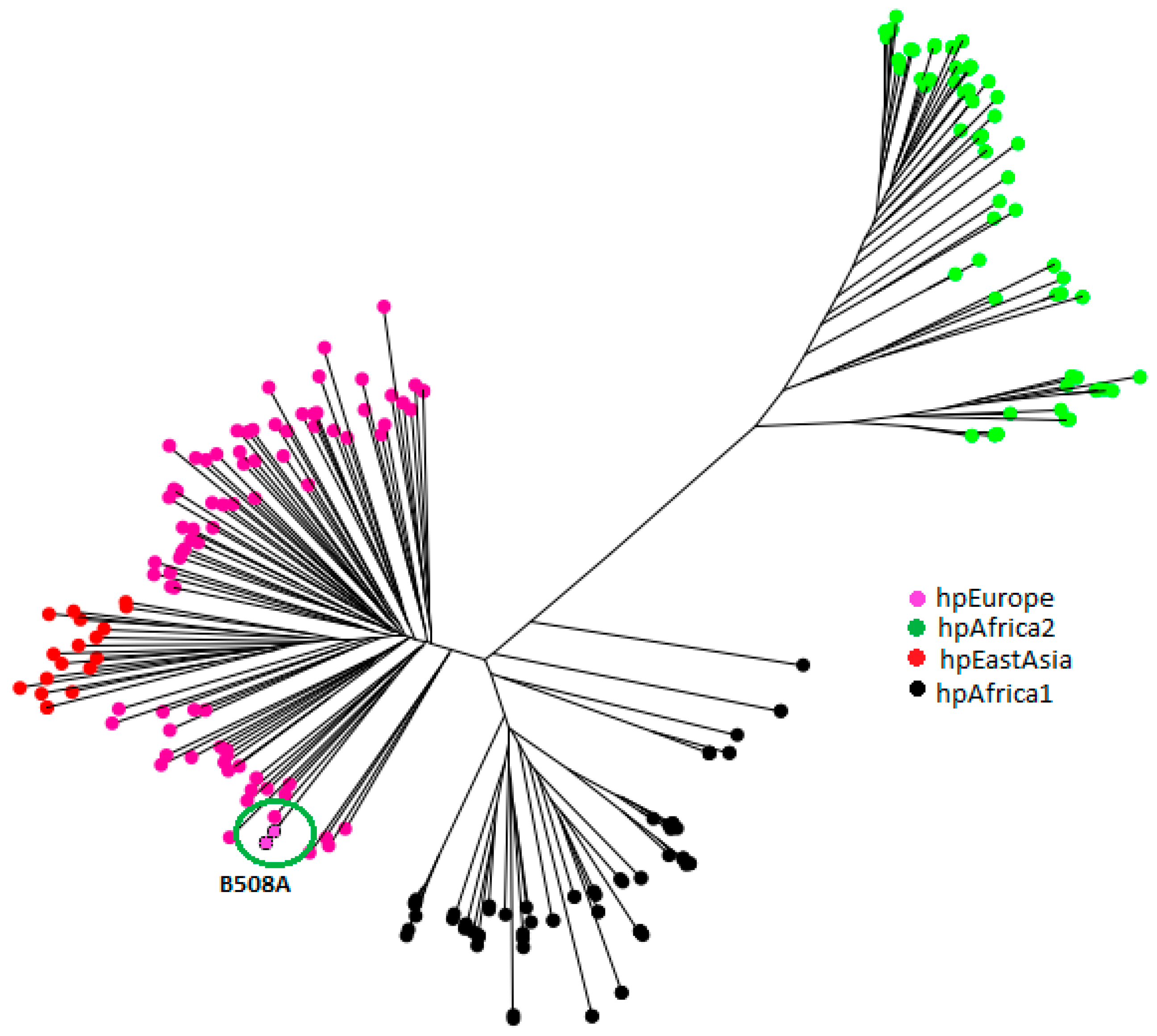
2.3. H. pylori Genetic Population
3. Discussion
4. Materials and Methods
4.1. H. pylori Strains and Genomic DNA Extraction and Sequencing
4.2. Genomic Comparison and Characterization
4.3. Virulence and Antimicrobial Resistance Genes
4.4. H. pylori Genetic Population Assignment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baltrus, D.A.; Blaser, M.J.; Guillemin, K. Helicobacter pylori genome plasticity. Genome Dyn. 2009, 6, 75–90. [Google Scholar] [CrossRef]
- Chiurillo, M.A.; Moran, Y.; Cañas, M.; Valderrama, E.; Granda, N.; Sayegh, M.; Ramírez, J.L. Genotyping of Helicobacter pylori virulence-associated genes shows high diversity of strains infecting patients in Western Venezuela. Int. J. Infect. Dis. 2013, 17, e750–e756. [Google Scholar] [CrossRef]
- Gressmann, H.; Linz, B.; Ghai, R.; Pleissner, K.-P.; Schlapbach, R.; Yamaoka, Y.; Kraft, C.; Suerbaum, S.; Meyer, T.F.; Achtman, M. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 2005, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; He, L.; Zhang, M.; Zhang, J. Comparative genomics of a Helicobacter pylori isolate from a Chinese Yunnan Naxi ethnic aborigine suggests high genetic divergence and phage insertion. PLoS ONE 2015, 10, e0120659. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.M.; Lu, Q.F.; Li, S.B.; Wang, J.P.; Chen, Y.L.; Huang, Y.Q.; Bi, H.K. Comparative genomics of H. pylori and non-pylori Helicobacter species to identify new regions associated with its pathogenicity and adaptability. Biomed. Res. Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Junaid, M.; Linn, A.K.; Javadi, M.B.; Al-Gubare, S.; Ali, N.; Katzenmeier, G. Vacuolating cytotoxin A (VacA)—A multi-talented pore-forming toxin from Helicobacter pylori. Toxicon 2016, 118, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.C.; Cao, P.; Peek, R.M.; Tummuru, M.K.R.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed]
- Rudi, J.; Kolb, C.; Maiwald, M.; Kuck, D.; Sieg, A.; Galle, P.R.; Stremmel, W. Diversity of Helicobacter pylori vacA and cagA genes and relationship to VacA and CagA protein expression, cytotoxin production, and associated disease. J. Clin. Microbiol. 1998, 36, 944–948. [Google Scholar] [CrossRef]
- Rhead, J.L.; Letley, D.P.; Mohammadi, M.; Hussein, N.; Mohagheghi, M.A.; Eshagh Hosseini, M.; Atherton, J.C. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007, 133, 926–936. [Google Scholar] [CrossRef]
- Mascellino, M.T.; Margani, M.; Oliva, A. Helicobacter pylori: Determinant and markers of virulence. Dis. Markers 2009, 27, 137–156. [Google Scholar] [CrossRef]
- Peek, R.M.; Thompson, S.A.; Atherton, J.C.; Blaser, M.J.; Miller, G.G. Expression of a novel ulcer-associated H. pylori gene, iceA, following adherence to gastric epithelial cells. Gastroenterology 1996, 110, A225. [Google Scholar]
- da Costa, D.M.; dos Santos Pereira, E.; Rabenhorst, S.H.B. What exists beyond cagA and vacA? Helicobacter pylori genes in gastric diseases. World J. Gastroenterol. 2015, 21, 10563–10572. [Google Scholar] [CrossRef]
- Piqué, N.; Palau, M.; Berlanga, M.; Miñana-Galbis, D. Advances in the research of new genetic markers for the detection of Helicobacter pylori infection. In Recent Advances in Pharmaceutical Sciences VI; Transworld Research Network: Kerala, India, 2016; pp. 165–188. Available online: http://diposit.ub.edu/dspace/bitstream/2445/103042/1/RecentAdvancesVI-1.pdf (accessed on 15 June 2019).
- Sycuro, L.K.; Pincus, Z.; Gutierrez, K.D.; Biboy, J.; Stern, C.A.; Vollmer, W.; Salama, N.R. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell 2010, 141, 822–933. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liao, T.; Debowski, A.W.; Tang, H.; Nilsson, H.O.; Stubbs, K.A.; Marshall, B.J.; Benghezal, M. Lipopolysaccharide structure and biosynthesis in Helicobacter pylori. Helicobacter 2016, 21, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Mathivanan, N.; Goyal, A. Bacterial adhesins, the pathogenic weapons to trick host defense arsenal. Biomed. Pharmacother. 2017, 93, 763–771. [Google Scholar] [CrossRef]
- Israel, D.A.; Peek, R.M. Surreptitious manipulation of the human host by Helicobacter pylori. Gut Microbes 2010, 1, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Teymournejad, O.; Mobarez, A.M.; Hassan, Z.M.; Talebi Bezmin Abadi, A. Binding of the Helicobacter pylori OipA causes apoptosis of host cells via modulation of Bax/Bcl-2 levels. Sci. Rep. 2017, 7, 8036. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfad, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Mobley, H.L.T.; Mendz, G.L.; Hazell, S.L. Helicobacter pylori: Physiology and Genetics; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Backert, S.; Clyne, M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2011, 16, 19–25. [Google Scholar] [CrossRef]
- Ali, A.; Naz, A.; Soares, S.C.; Bakhtiar, M.; Tiwari, S.; Hassan, S.S.; Hanan, F.; Ramos, R.; Pereira, U.; Barh, D.; et al. Pan-genome analysis of human gastric pathogen H. pylori: Comparative genomics and pathogenomics approaches to identify regions associated with pathogenicity and prediction of potential core therapeutic targets. Biomed. Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef]
- Kawai, M.; Furuta, Y.; Yahara, K.; Tsuru, T.; Oshima, K.; Handa, N.; Takahashi, N.; Yoshida, M.; Azuma, T.; Hattori, M.; et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 2011, 11, 104. [Google Scholar] [CrossRef]
- Cao, Q.; Didelot, X.; Wu, Z.; Li, Z.; He, L.; Li, Y.; Ni, M.; You, Y.; Lin, X.; Li, Z.; et al. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut 2015, 64, 554–561. [Google Scholar] [CrossRef]
- Palau, M.; Piqué, N.; Comeau, A.M.; Douglas, G.M.; Ramírez-Lázaro, M.J.; Lario, S.; Calvet, X.; Langille, M.G.I.; Miñana-Galbis, D. Detection of Helicobacter pylori microevolution and multiple infection from gastric biopsies by housekeeping gene amplicon sequencing. Pathogens 2020, 9, 97. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Palau, M.; Kulmann, M.; Ramírez-Lázaro, M.J.; Lario, S.; Quílez, M.E.; Campo, R.; Piqué, N.; Calvet, X.; Miñana-Galbis, D. Usefulness of housekeeping genes for the diagnosis of Helicobacter pylori infection, strain discrimination and detection of multiple infection. Helicobacter 2016, 21, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Weinert, L.A.; Welch, J.J. Why might bacterial pathogens have small genomes? Trends Ecol. Evol. 2017, 32, 936–947. [Google Scholar] [CrossRef]
- Brodsky, I.E.; Medzhitov, R. Targeting of immune signalling networks by bacterial pathogens. Nat. Cell. Biol. 2009, 11, 521–526. [Google Scholar] [CrossRef]
- Schulte, R.D.; Makus, C.; Schulenburg, H. Host-parasite coevolution favours parasite genetic diversity and horizontal gene transfer. J. Evol. Biol. 2013, 26, 1836–1840. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.L.; Hansen, L.M.; Bernick, D.L.; Abedrabbo, S.; Underwood, J.G.; Kong, N.; Huang, B.C.; Weis, A.M.; Weimer, B.C.; van Vliet, A.H.M.; et al. Fallacy of the unique genome: Sequence diversity within single Helicobacter pylori strains. MBio 2017, 8, e02321-16. [Google Scholar] [CrossRef]
- Fischer, W.; Buhrdorf, R.; Gerland, E.; Haas, R. Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect. Immun. 2001, 69, 6769–6775. [Google Scholar] [CrossRef] [PubMed]
- Ershova, A.S.; Rusinov, I.S.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry (Mosc.) 2015, 80, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.; Windsor, H.M.; Gajewski, J.P.; Hake, C.M.; Drautz, D.I.; Schuster, S.C.; Marshall, B.J. Helicobacter pylori genomic microevolution during naturally occurring transmission between adults. PLoS ONE 2013, 8, e82187. [Google Scholar] [CrossRef] [PubMed]
- Gu, H. Role of flagella in the pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef]
- Leker, K.; Lozano-Pope, I.; Bandyopadhyay, K.; Choudhury, B.P.; Obonyo, M. Comparison of lipopolysaccharides composition of two different strains of Helicobacter pylori. BMC Microbiol. 2017, 17, 226. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-J.; Blanke, S.R. Remodeling the host environment: Modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell. Infect. Microbiol. 2012, 2, 37. [Google Scholar] [CrossRef]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Su, Y.-L.; Huang, H.-L.; Huang, B.-S.; Chen, P.-C.; Chen, C.-S.; Wang, H.-L. Combination of OipA, BabA, and SabA as candidate biomarkers for predicting Helicobacter pylori-related gastric cancer. Sci. Rep. 2016, 6, 36442. [Google Scholar] [CrossRef]
- Berthenet, E.; Yahara, K.; Thorell, K.; Pascoe, B.; Meric, G.; Mikhail, J.M.; Engstrand, L.; Enroth, H.; Burette, A.; Megraud, F.; et al. A GWAS on Helicobacter pylori strains points to genetic variants associated with gastric cancer risk. BMC Biol. 2018, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Pormohammad, A.; Ghotaslo, R.; Leylabadlo, H.E.; Nasiri, M.J.; Dabiri, H.; Hashemi, A. Risk of gastric cancer in association with Helicobacter pylori different virulence factors: A systematic review and meta-analysis. Microb. Pathog. 2018, 118, 214–219. [Google Scholar] [CrossRef]
- García-Zea, J.A.; de la Herrán, R.; Rodríguez, F.R.; Navajas-Pérez, R.; Rejón, C.R. Detection and variability analyses of CRISPR-like loci in the H. pylori genome. PeerJ. 2019, 7, e6221. [Google Scholar] [CrossRef]
- Alba, C.; Blanco, A.; Alarcón, T. Antibiotic resistance in Helicobacter pylori. Curr. Opin. Infect. Dis. 2017, 30, 489–497. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf?ua=1 (accessed on 23 December 2020).
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–13403. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads. Available online: https://github.com/tseemann/snippy (accessed on 23 December 2020).
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief Bioinform. 2017, bbx081. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Lario, S.; Ramírez-Lázaro, M.J.; Aransay, A.M.; Lozano, J.J.; Montserrat, A.; Casalots, Á.; Junquera, F.; Álvarez, J.; Segura, F.; Campo, R.; et al. MicroRNA profiling in duodenal ulcer disease caused by Helicobacter pylori infection in a Western population. Clin. Microbiol. Infect. 2012, 18, E273–E282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Atherton, J.C.; Cover, T.L.; Twells, R.J.; Morales, M.R.; Hawkey, C.J.; Blaser, M.J. Simple and accurate PCR-based system for typing vacuolating cytotoxin alleles of Helicobacter pylori. J. Clin. Microbiol. 1999, 37, 2979–2982. [Google Scholar] [CrossRef]
- Edgar, R.C. PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinform. 2007, 8, 18. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Gerrits, M.M.; Godoy, A.P.; Kuipers, E.J.; Ribeiro, M.L.; Stoof, J.; Mendonça, S.; van Vliet, A.H.; Pedrazzoli, J. Jr.; Kusters, J.G. Multiple mutations in or adjacent to the conserved penicillin-binding protein motifs of the penicillin-binding protein 1A confer amoxicillin resistance to Helicobacter pylori. Helicobacter 2006, 11, 181–187. [Google Scholar] [CrossRef]
- Rimbara, E.; Noguchi, N.; Kawai, T.; Sasatsu, M. Mutations in penicillin-binding proteins 1, 2 and 3 are responsible for amoxicillin resistance in Helicobacter pylori. J. Antimicrob. Chemother. 2008, 61, 995–998. [Google Scholar] [CrossRef]
- Nishizawa, T.; Suzuki, H.; Tsugawa, H.; Muraoka, H.; Matsuzaki, J.; Hirata, K.; Ikeda, F.; Takahashi, M.; Hibi, T. Enhancement of amoxicillin resistance after unsuccessful Helicobacter pylori eradication. Antimicrob. Agents Chemother. 2011, 55, 3012–3014. [Google Scholar] [CrossRef]
- Stone, G.G.; Shortridge, D.E.E.; Versalovic, J.; Beyer, J.; Flamm, R.K.; Graham, D.Y.; Ghoneim, A.T.; Tanaka, S.K. A PCR-Oligonucleotide ligation assay to determine the prevalence of 23S rRNA gene mutations in clarithromycin-resistant Helicobacter pylori. Antimicrob. Agents Chemother. 1997, 41, 712–714. [Google Scholar] [CrossRef]
- Furuta, T.; Soya, Y.; Sugimoto, M.; Shirai, N.; Nakamura, A.; Kodaira, C.; Nishino, M.; Okuda, M.; Okimoto, T.; Murakami, K.; et al. Modified allele-specific primer–polymerase chain reaction method for analysis of susceptibility of Helicobacter pylori strains to clarithromycin. J. Gastroenterol. Hepatol. 2007, 22, 1810–1815. [Google Scholar] [CrossRef] [PubMed]
- Agudo, S.; Pérez-Pérez, G.; Alarcón, T.; López-Brea, M. Rapid detection of clarithromycin resistant Helicobacter pylori strains in Spanish patients by polymerase chain reaction-restriction fragment length polymorphism. Rev. Española Quimioter. 2011, 24, 32–36. Available online: https://seq.es/seq/0214-3429/24/1/agudo.pdf (accessed on 16 May 2019).
- Mahachai, V.; Sirimontaporn, N.; Tumwasorn, S.; Thong-Ngam, D.; Vilaichone, R.-K. Sequential therapy in clarithromycin-sensitive and -resistant Helicobacter pylori based on polymerase chain reaction. J. Gastroenterol. Hepatol. 2011, 26, 825–828. [Google Scholar] [CrossRef]
- Glocker, E.; Berning, M.; Gerrits, M.M.; Kusters, J.G.; Kist, M. Real-Time PCR screening for 16S rRNA mutations associated with resistance to tetracycline in Helicobacter pylori. Antimicrob. Agents Chemother. 2005, 49, 3166–3170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lawson, A.J.; Elviss, N.C.; Owen, R.J. Real-time PCR detection and frequency of 16S rDNA mutations associated with resistance and reduced susceptibility to tetracycline in Helicobacter pylori from England and Wales. J. Antimicrob. Chemother. 2005, 56, 282–286. [Google Scholar] [CrossRef]
- Domanovich-Asor, T.; Motro, Y.; Khalfin, B.; Craddock, H.A.; Peretz, A.; Moran-Gilad, J. Genomic analysis of antimicrobial resistance genotype-to-phenotype agreement in Helicobacter pylori. Microorganisms 2020, 9, 2. [Google Scholar] [CrossRef]
- Kwon, D.; El-Zaatari, F.A.K.; Kato, M.; Osato, M.S.; Reddy, R.; Yamaoka, Y.; Graham, D.Y. Analysis of rdxA and involvement of additional genes encoding NAD(P)H flavin oxidoreductase (FrxA) and ferredoxin-like protein (FdxB) in metronidazole resistance of Helicobacter pylori. Antimicrob. Agents Chemother. 2000, 44, 2133–2142. [Google Scholar] [CrossRef]
- Binh, T.T.; Suzuki, R.; Huyen, T.; Kwon, H.; Yamaoka, Y. Search for novel candidate mutations for metronidazole resistance in Helicobacter pylori using next-generation sequencing. Antimicrob. Agents Chemother. 2015, 59, 2343–2348. [Google Scholar] [CrossRef]
- Trespalacios-Rangél, A.A.; Otero, W.; Arévalo-Galvis, A.; Poutou-Piñales, R.A.; Rimbara, E.; Graham, D.Y. Surveillance of levofloxacin resistance in Helicobacter pylori isolates in Bogotá-Colombia (2009–2014). PLoS ONE 2016, 11, 1–10. [Google Scholar] [CrossRef]
- Zerbetto De Palma, G.; Mendiondo, N.; Wonaga, A.; Viola, L.; Ibarra, D.; Campitelli, E.; Salim, N.; Corti, R.; Goldman, C.; Catalano, M. Occurrence of mutations in the antimicrobial target genes related to levofloxacin, clarithromycin, and amoxicillin resistance in Helicobacter pylori. Microb. Drug Resist. 2017, 23, 351–358. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genome Characteristics | B508A-S1 | B508A-T2A | B508A-T4 |
|---|---|---|---|
| GC% | 39.0 | 39.0 | 38.9 |
| Genome Size (bp) | 1,586,749 | 1,584,784 | 1,585,256 |
| N50 | 200,200 | 106,266 | 124,923 |
| Genes (total) | 1577 | 1577 | 1581 |
| Genes (unique) | 2 | 13 | 3 |
| CDS (total) | 1535 | 1535 | 1536 |
| CDS (coding) | 1445 | 1442 | 1444 |
| rRNAs (5S, 16S, 23S) | 1, 1, 1 | 1, 1, 1 | 2, 1 1, 1 |
| tRNAs | 36 | 36 | 36 |
| ncRNAs | 3 | 3 | 3 |
| Pseudo Genes | 90 | 93 | 92 |
| vacA (PCR) | s1-s2/m1-m2 | s1-s2/m1-m2 | s1-s2/m1-m2 |
| vacA (genome) | s2/m2/i2 | s1/m2/i2 | s2/m2/i2 |
| cagA | - | - | - |
| GenBank Accession | QDJN00000000 | QDJM00000000 | QDJL00000000 |
| Type | Number of Polymorphisms | ||
|---|---|---|---|
| B508A-S1 Versus -T4 | B508A-S1 Versus -T2 | B508A-T2 Versus -T4 | |
| SNP 1 | 113 | 2454 | 2418 |
| MNP 2 | 10 | 198 | 201 |
| Complex | 29 | 618 | 619 |
| Deletions | 12 | 35 | 38 |
| Insertions | 5 | 39 | 40 |
| Total | 169 | 3344 | 3316 |
| Virulence Factors | Related Genes | B508A-S1 | B508A-T2A | B508A-T4 |
|---|---|---|---|---|
| Urease | 1 | 1 | 1 | 1 |
| Flagella | 2 | 1 | 1 | 1 |
| Lipopolysaccharide Lewis antigens | futA | 1 | 2 | 1 |
| futB | 1 | - | 1 | |
| futC | 2 | 2 | 1 | |
| Neutrophil-activating protein | napA | 1 | 1 | 1 |
| oipA/hopH | 1 | 1 | 1 | |
| Vacuolating cytotoxin | vacA | 1 | 1 | 1 |
| Adherence-associated lipoproteins | alpA/hopC | 1 | 1 | 1 |
| alpB/hopB | 1 | 1 | 1 | |
| Blood group antigen binding adhesins | babA/hopS | - | - | - |
| babB/hopT | 3 | 2 | 3 | |
| H. pylori adhesin A | hpaA | 1 | 1 | 1 |
| HopZ | hopZ | 1 | 1 | 1 |
| HorB | horB | 1 | 1 | 1 |
| Sialic acid binding proteins | sabA/hopP | 1 | 1 | 1 |
| sabB/hopO | - | - | - | |
| Duodenal ulcer promoting | dupA | 1 | 1 | 1 |
| Induced by contact with epithelium A 3 | iceA | 1 | 1 | 1 |
| Gene | Mutational Position | Susceptible Strains | B508A-S1 | B508A-T2 | B508A-T4 |
|---|---|---|---|---|---|
| pbp1 | V374L | V | V | V | V |
| E406A | E | E | E | E | |
| S414R | S | S | S | S | |
| T593A | T | T | T | T | |
| A599G | A | A | A | A | |
| V601G | V | V | V | V | |
| 23S rRNA | A2143G | A | A | A | A |
| A2144G | A | A | A | A | |
| T2183C | T | T | T | T | |
| C2196T | C | C | C | C | |
| A2224G | A | A | A | A | |
| 16S rRNA | A926T | A | A | A | A |
| G927T | G | G | G | G | |
| A928C | A | A | A | A | |
| frxA | A32V | A | A | A | A |
| A70T | A | A | A | A | |
| F72S | F | F | F | F | |
| G73S | G | G | G | G | |
| A138V | A | A | A | A | |
| A152V | A | A | A | A | |
| A153V | A | A | A | A | |
| C193S | C | C | C | C | |
| rdxA | G3A | G | G | G | G |
| C46T | C | C | C | C | |
| G238A | G | G | G | G | |
| G352A | G | G | G | G | |
| gyrA | D86N | D | D | D | D |
| N87K\I\Y | N | N | N | N | |
| A88V | A | A | A | A | |
| D91Y\G\N | D | D | D | D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palau, M.; Piqué, N.; Ramírez-Lázaro, M.J.; Lario, S.; Calvet, X.; Miñana-Galbis, D. Whole-Genome Sequencing and Comparative Genomics of Three Helicobacter pylori Strains Isolated from the Stomach of a Patient with Adenocarcinoma. Pathogens 2021, 10, 331. https://doi.org/10.3390/pathogens10030331
Palau M, Piqué N, Ramírez-Lázaro MJ, Lario S, Calvet X, Miñana-Galbis D. Whole-Genome Sequencing and Comparative Genomics of Three Helicobacter pylori Strains Isolated from the Stomach of a Patient with Adenocarcinoma. Pathogens. 2021; 10(3):331. https://doi.org/10.3390/pathogens10030331
Chicago/Turabian StylePalau, Montserrat, Núria Piqué, M. José Ramírez-Lázaro, Sergio Lario, Xavier Calvet, and David Miñana-Galbis. 2021. "Whole-Genome Sequencing and Comparative Genomics of Three Helicobacter pylori Strains Isolated from the Stomach of a Patient with Adenocarcinoma" Pathogens 10, no. 3: 331. https://doi.org/10.3390/pathogens10030331
APA StylePalau, M., Piqué, N., Ramírez-Lázaro, M. J., Lario, S., Calvet, X., & Miñana-Galbis, D. (2021). Whole-Genome Sequencing and Comparative Genomics of Three Helicobacter pylori Strains Isolated from the Stomach of a Patient with Adenocarcinoma. Pathogens, 10(3), 331. https://doi.org/10.3390/pathogens10030331