
Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Whole Genome Sequencing and Species Identification
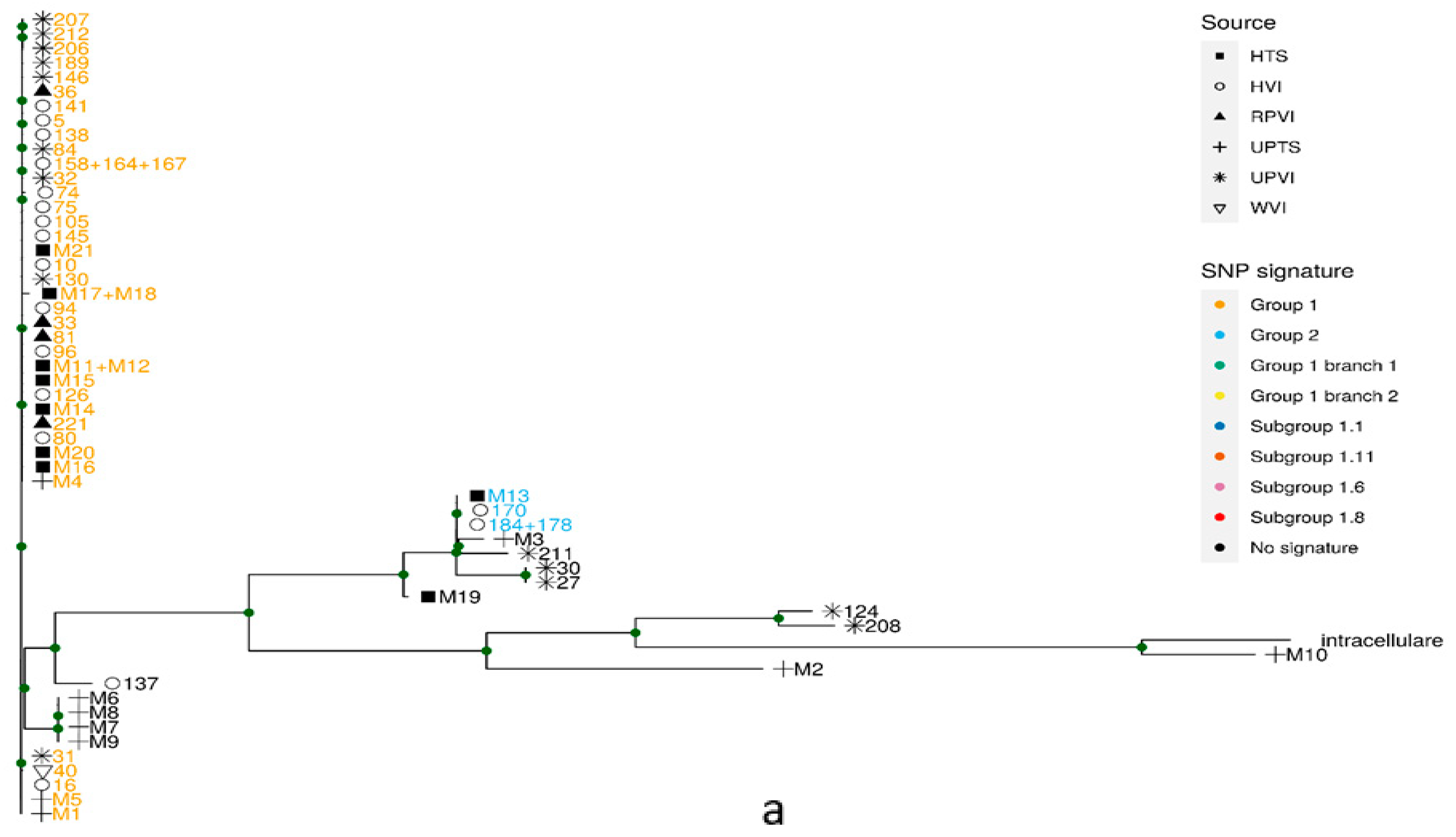
2.2. Identification of Known Phylogenetic Groups by Specific SNP Signatures
2.3. Phylogeny
2.4. Validation of the Results
3. Discussion
4. Materials and Methods
4.1. Bacterial Sampling
4.2. DNA Extraction and Species Identification by Hain Lifescience Assay Kit
4.3. Genomic DNA Isolation
4.4. Whole Genome Sequencing
4.5. Metagenomic Taxon Annotation of Reads
4.6. Metagenomic Binning
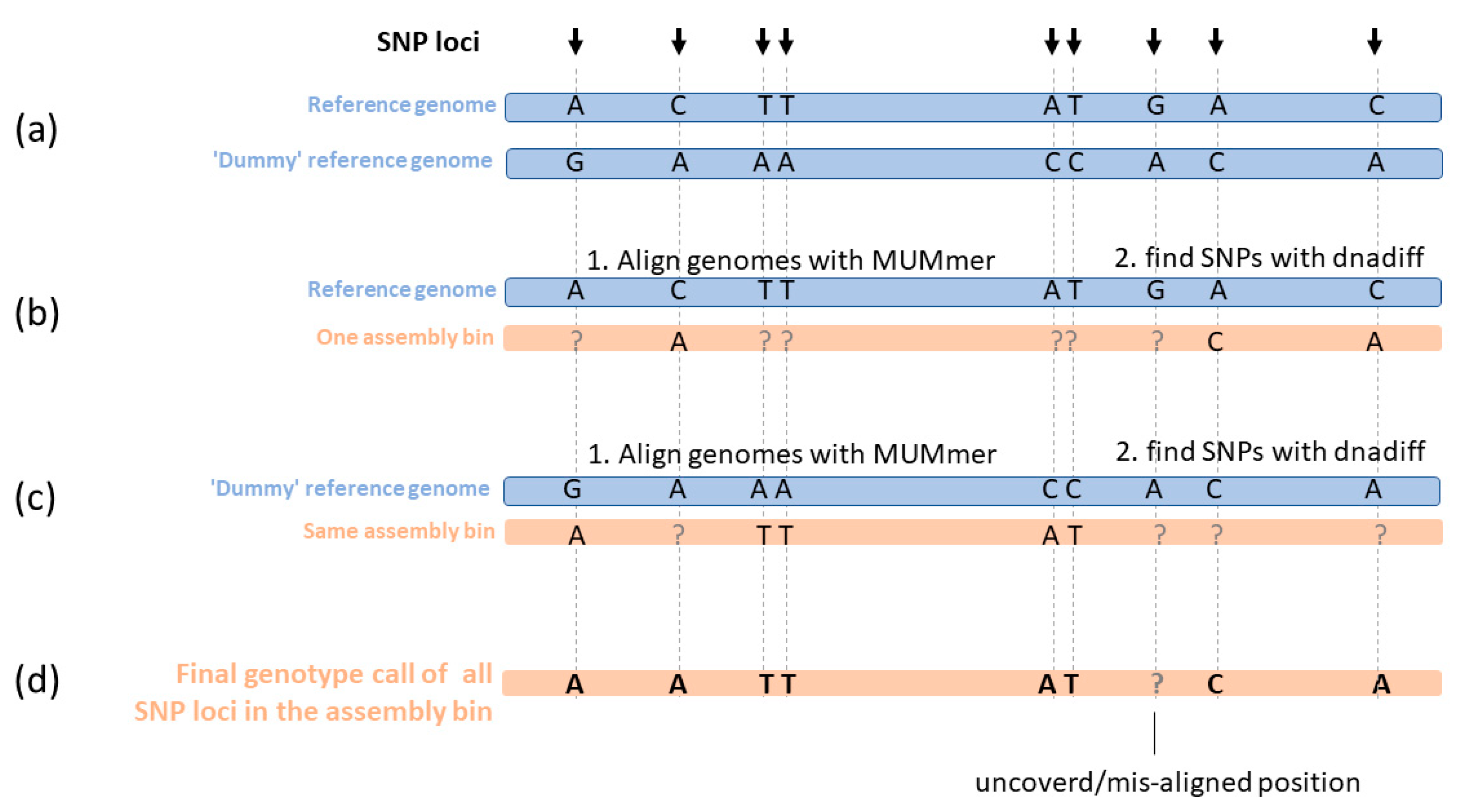
4.7. SNP Calling and Genotype Calling
4.8. SNP Based Phylogenetic Tree
4.9. Group Specific SNP Signatures
4.10. Group Specific Signature SNP in Clinical Samples, Using Mapping of Reads to Referecne Genome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Larsson, L.-O.; Polverino, E.; Hoefsloot, W.; Codecasa, L.R.; Diel, R.; Jenkins, S.G.; Loebinger, M.R. Pulmonary disease by non-tuberculous mycobacteria-clinical management, unmet needs and future perspectives. Expert Rev. Respir. Med. 2017, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Saddi, V.; Britton, P.N.; Selvadurai, H.; Robinson, P.D.; Pandit, C.; Marais, B.J.; Fitzgerald, D.A. Disease caused by non-tuberculous mycobacteria in children with cystic fibrosis. Paediatr. Respir. Rev. 2019, 29, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Misch, E.A.; Saddler, C.; Davis, J.M. Skin and Soft Tissue Infections Due to Nontuberculous Mycobacteria. Curr. Infect. Dis. Rep. 2018, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Ratnatunga, C.N.; Lutzky, V.P.; Kupz, A.; Doolan, D.L.; Reid, D.W.; Field, M.; Bell, S.C.; Thomson, R.M.; Miles, J.J. The Rise of Non-Tuberculosis Mycobacterial Lung Disease. Front. Immunol. 2020, 11, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saifi, M.; Jabbarzadeh, E.; Bahrmand, A.R.; Karimi, A.; Pourazar, S.; Fateh, A.; Masoumi, M.; Vahidi, E. HSP65-PRA identification of non-tuberculosis mycobacteria from 4892 samples suspicious for mycobacterial infections. Clin. Microbiol. Infect. 2013, 19, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Araj, G.F.; Baba, O.Z.; Itani, L.Y.; Avedissian, A.Z.; Sobh, G.M. Non-tuberculous mycobacteria profiles and their anti-mycobacterial resistance at a major medical center in Lebanon. J. Infect. Dev. Ctries. 2019, 13, 612–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, F.P.; Pohle, P.; Kernbach, M.; Sievert, D.; Hillemann, D.; Rupp, J.; Hombach, M.; Kranzer, K. Differential drug susceptibility patterns of Mycobacterium chimaera and other members of the Mycobacterium avium-intracellulare complex. Clin. Microbiol. Infect. 2019, 25, 379e1–379e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouioui, I.; Carro, L.; Garcia-Lopez, M.; Meier-Kolthoff, J.P.; Woyke, T.; Kyrpides, N.C.; Pukall, R.; Klenk, H.P.; Goodfellow, M.; Goker, M. Genome-Based Taxonomic Classification of the Phylum Actinobacteria. Front. Microbiol. 2018, 9, 2007. [Google Scholar] [CrossRef] [Green Version]
- Van Ingen, J.; Turenne, C.Y.; Tortoli, E.; Wallace, R.J., Jr.; Brown-Elliott, B.A. A definition of the Mycobacterium avium complex for taxonomical and clinical purposes, a review. Int. J. Syst. Evol. Microbiol. 2018, 68, 3666–3677. [Google Scholar] [CrossRef]
- Achermann, Y.; Rossle, M.; Hoffmann, M.; Deggim, V.; Kuster, S.; Zimmermann, D.R.; Bloemberg, G.; Hombach, M.; Hasse, B. Prosthetic valve endocarditis and bloodstream infection due to Mycobacterium chimaera. J. Clin. Microbiol. 2013, 51, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Kohler, P.; Kuster, S.P.; Bloemberg, G.; Schulthess, B.; Frank, M.; Tanner, F.C.; Rossle, M.; Boni, C.; Falk, V.; Wilhelm, M.J.; et al. Healthcare-associated prosthetic heart valve, aortic vascular graft, and disseminated Mycobacterium chimaera infections subsequent to open heart surgery. Eur. Heart. J. 2015, 36, 2745–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, K.M.; Lawsin, A.; Hasan, N.A.; Strong, M.; Halpin, A.L.; Rodger, R.R.; Moulton-Meissner, H.; Crist, M.B.; Schwartz, S.; Marders, J.; et al. Notes from the Field: Mycobacterium chimaera Contamination of Heater-Cooler Devices Used in Cardiac Surgery-United States. Morb. Morta.l Wkly. Rep. 2016, 65, 1117–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, D.; Howden, B.; Stinear, T. Mycobacterium chimaera Spread from Heating and Cooling Units in Heart Surgery. N. Engl. J. Med. 2017, 376, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, R.; Banderet, F.; Egli, A.; Keller, P.M.; Frei, R.; Dobele, T.; Eckstein, F.; Widmer, A.F. Different Types of Heater-Cooler Units and Their Risk of Transmission of Mycobacterium chimaera During Open-Heart Surgery: Clues From Device Design. Infect. Control Hosp. Epidemiol. 2018, 39, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.; Holler, C.; Jacobshagen, A.; Hamouda, O.; Abu Sin, M.; Monnet, D.L.; Plachouras, D.; Eckmanns, T. Contamination during production of heater-cooler units by Mycobacterium chimaera potential cause for invasive cardiovascular infections: Results of an outbreak investigation in Germany, April 2015 to February 2016. Euro. Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ingen, J.; Kohl, T.A.; Kranzer, K.; Hasse, B.; Keller, P.M.; Katarzyna Szafranska, A.; Hillemann, D.; Chand, M.; Schreiber, P.W.; Sommerstein, R.; et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: A molecular epidemiological study. Lancet Infect. Dis. 2017, 17, 1033–1041. [Google Scholar] [CrossRef]
- Hasan, N.A.; Epperson, L.E.; Lawsin, A.; Rodger, R.R.; Perkins, K.M.; Halpin, A.L.; Perry, K.A.; Moulton-Meissner, H.; Diekema, D.J.; Crist, M.B.; et al. Genomic Analysis of Cardiac Surgery-Associated Mycobacterium chimaera Infections, United States. Emerg. Infect. Dis. 2019, 25, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, E.; Jensen, E.T.; Rasmussen, E.M.; Folkvardsen, D.B.; Norman, A.; Lillebaek, T. Mycobacterium chimaera in Heater-Cooler Units in Denmark Related to Isolates from the United States and United Kingdom. Emerg. Infect. Dis. 2017, 23, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, J.; Feng, Y.; Wang, X.; McNally, A.; Zong, Z. Identification of Mycobacterium chimaera in heater-cooler units in China. Sci. Rep. 2018, 8, 7843. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Cule, M.L.; Griffiths, D.; Crook, D.W.; Peto, T.E.; Walker, A.S.; Wilson, D.J. Detection of mixed infection from bacterial whole genome sequence data allows assessment of its role in Clostridium difficile transmission. PLoS Comput. Biol. 2013, 9, e1003059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anyansi, C.; Keo, A.; Walker, B.J.; Straub, T.J.; Manson, A.L.; Earl, A.M.; Abeel, T. QuantTB-a method to classify mixed Mycobacterium tuberculosis infections within whole genome sequencing data. BMC Genomics 2020, 21, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Huang, L.; Shi, C.; Wang, L.; Yu, R. UltraStrain: An NGS-Based Ultra Sensitive Strain Typing Method for Salmonella enterica. Front. Genet. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Delmont, T.O.; Raguideau, S.; Alneberg, J.; Darling, A.E.; Collins, G.; Eren, A.M. DESMAN: A new tool for de novo extraction of strains from metagenomes. Genome Biol. 2017, 18, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kitajima, M.; Whittle, A.J.; Liu, W.T. Benefits of Genomic Insights and CRISPR-Cas Signatures to Monitor Potential Pathogens across Drinking Water Production and Distribution Systems. Front. Microbiol. 2017, 8, 2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chand, M.; Lamagni, T.; Kranzer, K.; Hedge, J.; Moore, G.; Parks, S.; Collins, S.; Del Ojo Elias, C.; Ahmed, N.; Brown, T.; et al. Insidious Risk of Severe Mycobacterium chimaera Infection in Cardiac Surgery Patients. Clin. Infect. Dis. 2017, 64, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scriven, J.E.; Scobie, A.; Verlander, N.Q.; Houston, A.; Collyns, T.; Cajic, V.; Kon, O.M.; Mitchell, T.; Rahama, O.; Robinson, A.; et al. Mycobacterium chimaera infection following cardiac surgery in the United Kingdom: Clinical features and outcome of the first 30 cases. Clin. Microbiol. Infect. 2018, 24, 1164–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjemian, J.; Olivier, K.N.; Seitz, A.E.; Holland, S.M.; Prevots, D.R. Prevalence of nontuberculous mycobacterial lung disease in U.S. Medicare beneficiaries. Am. J. Respir. Crit. Care. Med. 2012, 185, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Boyle, D.P.; Zembower, T.R.; Reddy, S.; Qi, C. Comparison of Clinical Features, Virulence, and Relapse among Mycobacterium avium Complex Species. Am. J. Respir. Crit. Care. Med. 2015, 191, 1310–1317. [Google Scholar] [CrossRef]
- Koh, W.-J.; Chang, B.; Jeong, B.-H.; Jeon, K.; Kim, S.-Y.; Lee, N.Y.; Ki, C.-S.; Kwon, O.J. Increasing Recovery of Nontuberculous Mycobacteria from Respiratory Specimens over a 10-Year Period in a Tertiary Referral Hospital in South Korea. Tuberc. Respir. Dis. 2013, 75, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, H. Mycobacterium avium complex infection mimicking lung cancer. BMJ Case Rep. 2019, 12, e228847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-Y.; Shin, S.H.; Moon, S.M.; Yang, B.; Kim, H.; Kwon, O.J.; Huh, H.J.; Ki, C.-S.; Lee, N.Y.; Shin, S.J.; et al. Distribution and clinical significance of Mycobacterium avium complex species isolated from respiratory specimens. Diagn. Microbiol. Infect. Dis. 2017, 88, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Rosero, C.I.; Shams, W.E. Mycobacterium chimaera infection masquerading as a lung mass in a healthcare worker. IDCases 2019, 15, e00526. [Google Scholar] [CrossRef] [PubMed]
- Kranzer, K.; Ahmed, N.; Hoffman, P.; Pink, F. Protocol for Environmental Sampling, Processing and Culturing of Water and Air Samples for the Isolation of Slow-Growing Mycobacteria Standard Operating Procedure. 2016. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/540325/Air_water_environmental_sampling_SOP_V2.pdf (accessed on 20 October 2021).
- Kent, P.T.; Kubica, G.P. Public Health Mycobacteriology: A Guide for the Level III Laboratory; Centers for Disease Control: Atlanta, GA, USA, 1985; p. 207.
- Freidlin, P.J.; Goldblatt, D.; Kaidar-Shwartz, H.; Rorman, E. Polymorphic exact tandem repeat A (PETRA): A newly defined lineage of mycobacterium tuberculosis in israel originating predominantly in Sub-Saharan Africa. J. Clin. Microbiol. 2009, 47, 4006–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample Number | Source 1 | DNA Extraction 2 | Illumina Sequencing Instrument 3 | PCR Based Identification 4 | Year of Sampling | HCU Production Date 5 | HCU Number | Medical Center 6 |
|---|---|---|---|---|---|---|---|---|
| M1 | cl.: sputum | RFLP | HiSeq | M. chimaera | 2017 | Tm | ||
| M2 | cl.: sputum | RFLP | HiSeq | M. chimaera | 2017 | Ma | ||
| M3 | cl.: sputum | RFLP | HiSeq | M. chimaera | 2017 | Abct | ||
| M4 | cl.: sputum | Magnapure | MiSeq | M. chimaera | 2017 | Tlm | ||
| M5 | cl.: sputum | Magnapure | MiSeq | M. chimaera | 2017 | Ma | ||
| M6 | cl.: sputum | Magnapure | MiSeq | M. chimaera | 2017 | Nt | ||
| M7 | cl.: sputum | RFLP | MiSeq | M. chimaera | 2017 | Rm | ||
| M8 | cl.: sputum | RFLP | MiSeq | M. chimaera | 2017 | Nt | ||
| M9 | cl.: thoracic biopsy | RFLP | MiSeq | M. chimaera | 2014 | - | ||
| M10 | cl.: pleural fluid | RFLP | HiSeq | M. intracellulare | 2013 | - | ||
| M11 | HCU | Magnapure | MiSeq | M. chimaera | 2017 | 2011 | 16S12916 | Lc |
| M12 | HCU | Magnapure | MiSeq | M. chimaera | 2017 | 2004 | 16S10462 | Lc |
| M13 | HCU | Magnapure | MiSeq | M. chimaera | 2017 | 16S12007 | Nk | |
| M14 | HCU | Magnapure | MiSeq | M. chimaera | 2017 | 2004 | 16S10468 | Ahs |
| M15 | HCU | Magnapure | MiSeq | M. chimaera | 2017 | 2011 | 16S12918 | Rs |
| M16 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 2007 | 16S10888 | Lc |
| M17 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 2004 | 16S10448 | Nw |
| M18 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 2007 | 16S10890 | As |
| M19 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 2004 | 16S10395 | Hh |
| M20 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 16S10082 | Ks | |
| M21 | HCU | RFLP | HiSeq | M. chimaera | 2017 | 2014 | 16S14090 | Aa |
| M22 | HCU | Magnapure | MiSeq | M. chimaera + M. gordonae | 2017 | 2015 | 16S15449 | Ap |
| M23 | HCU | Magnapure | MiSeq | M. chimaera + NTM | 2017 | 2015 | 16S15447 | Nw |
| Sample Number | Source 1 | PCR Based Identification 2* | Reads’ Most Abundant Species 3* | Reads’ M. chimaera Abundance 4* | Number of Bins 5 | Most Similar Genome to Bin (# K-mers) 6* |
|---|---|---|---|---|---|---|
| M1 | cl.: sputum | M. chimaera | M. chimaera (3.6%) | 3.6% | 1 | M. chimaera strain MCIMRL2 (920) |
| M2 | cl.: sputum | M. chimaera | M. chimaera (0.8%) | 0.8% | 1 | M. sp. TKK-01-0059 (874) |
| M3 | cl.: sputum | M. chimaera | M. chimaera (6.4%) | 6.4% | 1 | M. chimaera strain ZUERICH-2 (896) |
| M4 | cl.: sputum | M. chimaera | M. chimaera (4.4%) | 4.4% | 1 | M. chimaera strain ZUERICH-1 (805) |
| M5 | cl.: sputum | M. chimaera | M. chimaera (3.8%) | 3.8% | 1 | M. chimaera strain ZUERICH-1 (325) |
| M6 | cl.: sputum | M. chimaera | M. chimaera (4.6%) | 4.6% | 1 | M. chimaera strain DSM 44,623 (880) |
| M7 | cl.: sputum | M. chimaera | M. chimaera (4.4%) | 4.4% | 1 | M. chimaera strain DSM 44,623 (893) |
| M8 | cl.: sputum | M. chimaera | Shewanella decolorationis (5.6%) | 4.2% | 1 | M. chimaera strain DSM 44,623 (903) |
| M9 | cl.: thoracic biopsy | M. chimaera | Bacillus azotoformans (6.6%) | 3.9% | 1 | M. chimaera strain DSM 44,623 (873) |
| M10 | cl.: pleural fluid | M. intracellulare | M. intracellulare (6.2%) | 0.2% | 1 | M. intracellulare MIN_061107_1834 (801) |
| M11 | HCU | M. chimaera | M. chimaera (3.4%) | 3.4% | 1 | M. chimaera strain CDC 2015-22-71 (987) |
| M12 | HCU | M. chimaera | M. chimaera (3.6%) | 3.6% | 1 | M. chimaera strain CDC 2015-22-71 (987) |
| M13 | HCU | M. chimaera | M. chimaera (10.3%) | 10.3% | 1 | M. chimaera strain ZUERICH-2 (963) |
| M14 | HCU | M. chimaera | M. chimaera (4.7%) | 4.7% | 1 | M. chimaera strain ZUERICH-1 (1000) |
| M15 | HCU | M. chimaera | M. chimaera (4.1%) | 4.1% | 1 | M. chimaera strain ZUERICH-1 (999) |
| M16 | HCU | M. chimaera | M. gordonae (15.1%) | 0.6% | 2 | M. gordonae strain 1275229.4 (567) |
| M. chimaera strain ZUERICH-1 (900) | ||||||
| M17 | HCU | M. chimaera | M. gordonae (18.5%) | 0.2% | 2 | M. gordonae strain 1275229.4 (609) |
| M. chimaera strain WCHMC000032 (841) | ||||||
| M18 | HCU | M. chimaera | M. gordonae (17.7%) | 0.3% | 2 | M. gordonae strain 1275229.4 (609) |
| M. chimaera strain WCHMC000032 (841) | ||||||
| M19 | HCU | M. chimaera | M. gordonae (12.5%) | 0.2% | 2 | M. gordonae strain 1275229.4 (352) |
| M. chimaera strain SJ42 (743) | ||||||
| M20 | HCU | M. chimaera | M. sp. (6.8%) | 0.9% | 2 | M. sp. strain DS2.013 (767) |
| M. chimaera strain WCHMC000030 (897) | ||||||
| M21 | HCU | M. chimaera | M. gordonae (17%) | 0.3% | 2 | M. chimaera strain WCHMC000032 (867) |
| M. gordonae strain 1275229.4 (645) | ||||||
| M22 | HCU | M. chimaera + M. gordonae | M. gordonae (18.6%) | 0.9% | 1 | M. gordonae strain 1275229.4 (605) |
| M23 | HCU | M. chimaera + NTM | M. gordonae (16.6%) | 0.3% | 1 | M. gordonae strain 1275229.4 (562) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubinstein, M.; Grossman, R.; Nissan, I.; Schwaber, M.J.; Carmeli, Y.; Kaidar-Shwartz, H.; Dveyrin, Z.; Rorman, E. Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated. Pathogens 2021, 10, 1392. https://doi.org/10.3390/pathogens10111392
Rubinstein M, Grossman R, Nissan I, Schwaber MJ, Carmeli Y, Kaidar-Shwartz H, Dveyrin Z, Rorman E. Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated. Pathogens. 2021; 10(11):1392. https://doi.org/10.3390/pathogens10111392
Chicago/Turabian StyleRubinstein, Mor, Rona Grossman, Israel Nissan, Mitchell J. Schwaber, Yehuda Carmeli, Hasia Kaidar-Shwartz, Zeev Dveyrin, and Efrat Rorman. 2021. "Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated" Pathogens 10, no. 11: 1392. https://doi.org/10.3390/pathogens10111392
APA StyleRubinstein, M., Grossman, R., Nissan, I., Schwaber, M. J., Carmeli, Y., Kaidar-Shwartz, H., Dveyrin, Z., & Rorman, E. (2021). Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated. Pathogens, 10(11), 1392. https://doi.org/10.3390/pathogens10111392