
The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
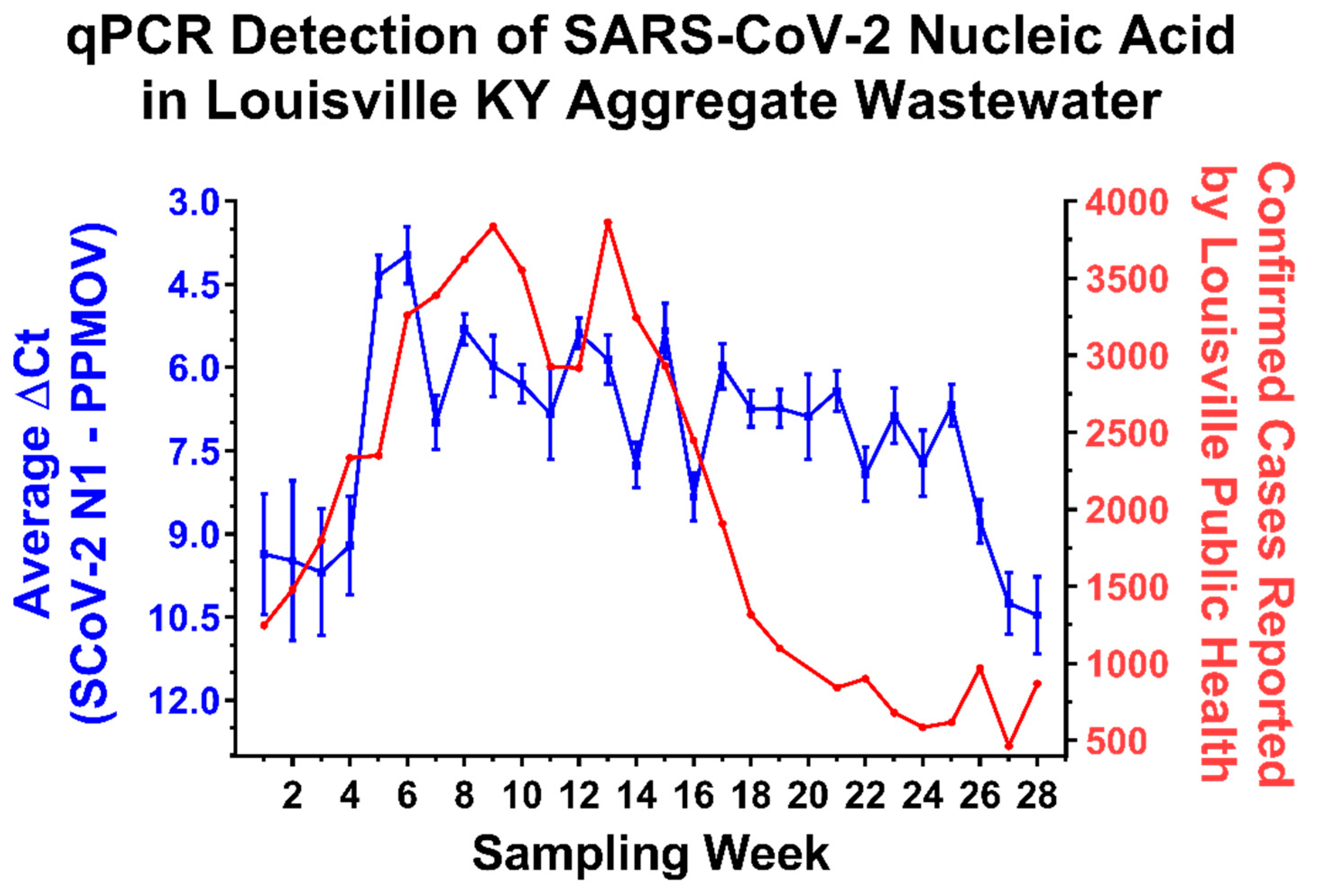
2.1. Detection of SARS-CoV-2 RNA in Aggregate Wastewater Samples Correlates with Community Prevalence
2.2. Aggregate Wastewater Sources Can Provide a Rapid Means to Detect the Prevalence of Variants of Interest/Concern
2.3. The Analysis of Aggregate Wastewater Sources Can Provide Broad Genetic Information on a Community-Level
3. Discussion
3.1. Aggregate Wastewater Analyses Are Indicative of Prevalence/Genetic Variation on a Community-Level
3.2. Sequence Analysis of SARS-CoV-2 Genetic Diversity Reveals Potential Biological Insights
4. Conclusions
5. Materials and Methods
5.1. Wastewater Sample Prep
5.2. cDNA Synthesis
5.3. Library Preparation
5.4. Next-Generation Sequencing
5.5. Data Analysis
5.6. Correlation/Statistical Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Achak, M.; Alaoui Bakri, S.; Chhiti, Y.; M’Hamdi Alaoui, F.E.; Barka, N.; Boumya, W. SARS-CoV-2 in hospital wastewater during outbreak of COVID-19: A review on detection, survival and disinfection technologies. Sci. Total Environ. 2021, 761, 143192. [Google Scholar] [CrossRef]
- Scott, L.C.; Aubee, A.; Babahaji, L.; Vigil, K.; Tims, S.; Aw, T.G. Targeted wastewater surveillance of SARS-CoV-2 on a university campus for COVID-19 outbreak detection and mitigation. Environ. Res. 2021, 200, 111374. [Google Scholar] [CrossRef] [PubMed]
- Spurbeck, R.R.; Minard-Smith, A.; Catlin, L. Feasibility of neighborhood and building scale wastewater-based genomic epidemiology for pathogen surveillance. Sci. Total Environ. 2021, 789, 147829. [Google Scholar] [CrossRef]
- Fernandez-Cassi, X.; Scheidegger, A.; Banziger, C.; Cariti, F.; Tunas Corzon, A.; Ganesanandamoorthy, P.; Lemaitre, J.C.; Ort, C.; Julian, T.R.; Kohn, T. Wastewater monitoring outperforms case numbers as a tool to track COVID-19 incidence dynamics when test positivity rates are high. Water Res. 2021, 200, 117252. [Google Scholar] [CrossRef]
- Roka, E.; Khayer, B.; Kis, Z.; Kovacs, L.B.; Schuler, E.; Magyar, N.; Malnasi, T.; Oravecz, O.; Palyi, B.; Pandics, T.; et al. Ahead of the second wave: Early warning for COVID-19 by wastewater surveillance in Hungary. Sci. Total Environ. 2021, 786, 147398. [Google Scholar] [CrossRef] [PubMed]
- Colosi, L.M.; Barry, K.E.; Kotay, S.M.; Porter, M.D.; Poulter, M.D.; Ratliff, C.; Simmons, W.; Steinberg, L.I.; Wilson, D.D.; Morse, R.; et al. Development of Wastewater Pooled Surveillance of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) from Congregate Living Settings. Appl. Environ. Microbiol. 2021, 87, e0043321. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Angel, N.; Edson, J.; Bibby, K.; Bivins, A.; O’Brien, J.W.; Choi, P.M.; Kitajima, M.; Simpson, S.L.; Li, J.; et al. First confirmed detection of SARS-CoV-2 in untreated wastewater in Australia: A proof of concept for the wastewater surveillance of COVID-19 in the community. Sci. Total Environ. 2020, 728, 138764. [Google Scholar] [CrossRef]
- Bivins, A.; North, D.; Ahmad, A.; Ahmed, W.; Alm, E.; Been, F.; Bhattacharya, P.; Bijlsma, L.; Boehm, A.B.; Brown, J.; et al. Wastewater-Based Epidemiology: Global Collaborative to Maximize Contributions in the Fight Against COVID-19. Environ. Sci. Technol. 2020, 54, 7754–7757. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kulandaivelu, J.; Zhang, S.; Shi, J.; Sivakumar, M.; Mueller, J.; Luby, S.; Ahmed, W.; Coin, L.; Jiang, G. Data-driven estimation of COVID-19 community prevalence through wastewater-based epidemiology. Sci. Total Environ. 2021, 789, 147947. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, X.; Li, S.; Lam, N.S.; Wang, Y.; Chu, D.K.W.; Poon, L.L.M.; Tun, H.M.; Peiris, M.; Deng, Y.; et al. The first case study of wastewater-based epidemiology of COVID-19 in Hong Kong. Sci. Total Environ. 2021, 790, 148000. [Google Scholar] [CrossRef]
- Hillary, L.S.; Farkas, K.; Maher, K.H.; Lucaci, A.; Thorpe, J.; Distaso, M.A.; Gaze, W.H.; Paterson, S.; Burke, T.; Connor, T.R.; et al. Monitoring SARS-CoV-2 in municipal wastewater to evaluate the success of lockdown measures for controlling COVID-19 in the UK. Water Res. 2021, 200, 117214. [Google Scholar] [CrossRef] [PubMed]
- Jafferali, M.H.; Khatami, K.; Atasoy, M.; Birgersson, M.; Williams, C.; Cetecioglu, Z. Benchmarking virus concentration methods for quantification of SARS-CoV-2 in raw wastewater. Sci. Total Environ. 2021, 755, 142939. [Google Scholar] [CrossRef] [PubMed]
- Weidhaas, J.; Aanderud, Z.T.; Roper, D.K.; VanDerslice, J.; Gaddis, E.B.; Ostermiller, J.; Hoffman, K.; Jamal, R.; Heck, P.; Zhang, Y.; et al. Correlation of SARS-CoV-2 RNA in wastewater with COVID-19 disease burden in sewersheds. Sci. Total Environ. 2021, 775, 145790. [Google Scholar] [CrossRef] [PubMed]
- Westhaus, S.; Weber, F.A.; Schiwy, S.; Linnemann, V.; Brinkmann, M.; Widera, M.; Greve, C.; Janke, A.; Hollert, H.; Wintgens, T.; et al. Detection of SARS-CoV-2 in raw and treated wastewater in Germany—Suitability for COVID-19 surveillance and potential transmission risks. Sci. Total Environ. 2021, 751, 141750. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, J.; Xiao, A.; Gu, X.; Lee, W.L.; Armas, F.; Kauffman, K.; Hanage, W.; Matus, M.; Ghaeli, N.; et al. SARS-CoV-2 Titers in Wastewater Are Higher than Expected from Clinically Confirmed Cases. Msystems 2020, 5, e00614-20. [Google Scholar] [CrossRef]
- Gonzalez, R.; Curtis, K.; Bivins, A.; Bibby, K.; Weir, M.H.; Yetka, K.; Thompson, H.; Keeling, D.; Mitchell, J.; Gonzalez, D. COVID-19 surveillance in Southeastern Virginia using wastewater-based epidemiology. Water Res. 2020, 186, 116296. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, J.L.; Rouchka, E.C.; Waigel, S.; Sokoloski, K.; Chung, D.; Zacharias, W.; Zhang, M.; Chariker, J.; Talley, D.; Santisteban, I.; et al. A rapid assessment of wastewater for genomic surveillance of SARS-CoV-2 variants at sewershed scale in Louisville, KY. medRxiv 2021. [Google Scholar] [CrossRef]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, M.; Chen, K.; Cheng, L.; Weng, T.; Shi, D.; et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e1289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Munnink, B.B.O.; Schapendonk, C.M.E.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and Diversity through Community Wastewater Sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 2021, 197, 117104. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Ramos-Mandujano, G.; Tian, Y.; Hala, S.; Xu, J.; Mfarrej, S.; Esteban, C.R.; Delicado, E.N.; Alofi, F.S.; Khogeer, A.; et al. Simultaneous Detection and Mutation Surveillance of SARS-CoV-2 and co-infections of multiple respiratory viruses by Rapid field-deployable sequencing. Med 2021, 2, 689–700.e4. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Kraberger, S.; Hadfield, J.; Driver, E.M.; Bowes, D.; Holland, L.A.; Faleye, T.O.C.; Adhikari, S.; Kumar, R.; Inchausti, R.; et al. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. Water Res. 2021. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. MBio 2021, 12, e02703-20. [Google Scholar] [CrossRef]
- Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098. [Google Scholar] [CrossRef]
- Yeager, R.; Holm, R.H.; Saurabh, K.; Fuqua, J.L.; Talley, D.; Bhatnagar, A.; Smith, T. Wastewater Sample Site Selection to Estimate Geographically Resolved Community Prevalence of COVID-19: A Sampling Protocol Perspective. Geohealth 2021, 5, e2021GH000420. [Google Scholar] [CrossRef]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2021, 2, e13–e22. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Huang, D.; Lee, C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W.; et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Zhao, S.; Wu, X.; Lan, W.; Ren, Y.; Cui, L.; Lan, Q.; et al. V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J. Virol. 2021, 95, JVI-00617. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.E.; Li, J.; Machamer, C.E. The cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds COPI and promotes interaction with membrane protein. J. Virol. 2007, 81, 2418–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rouchka, E.C.; Chariker, J.H.; Saurabh, K.; Waigel, S.; Zacharias, W.; Zhang, M.; Talley, D.; Santisteban, I.; Puccio, M.; Moyer, S.; et al. The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale. Pathogens 2021, 10, 1271. https://doi.org/10.3390/pathogens10101271
Rouchka EC, Chariker JH, Saurabh K, Waigel S, Zacharias W, Zhang M, Talley D, Santisteban I, Puccio M, Moyer S, et al. The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale. Pathogens. 2021; 10(10):1271. https://doi.org/10.3390/pathogens10101271
Chicago/Turabian StyleRouchka, Eric C., Julia H. Chariker, Kumar Saurabh, Sabine Waigel, Wolfgang Zacharias, Mei Zhang, Daymond Talley, Ian Santisteban, Madeline Puccio, Sarah Moyer, and et al. 2021. "The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale" Pathogens 10, no. 10: 1271. https://doi.org/10.3390/pathogens10101271
APA StyleRouchka, E. C., Chariker, J. H., Saurabh, K., Waigel, S., Zacharias, W., Zhang, M., Talley, D., Santisteban, I., Puccio, M., Moyer, S., Holm, R. H., Yeager, R. A., Sokoloski, K. J., Fuqua, J., Bhatnagar, A., & Smith, T. (2021). The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale. Pathogens, 10(10), 1271. https://doi.org/10.3390/pathogens10101271