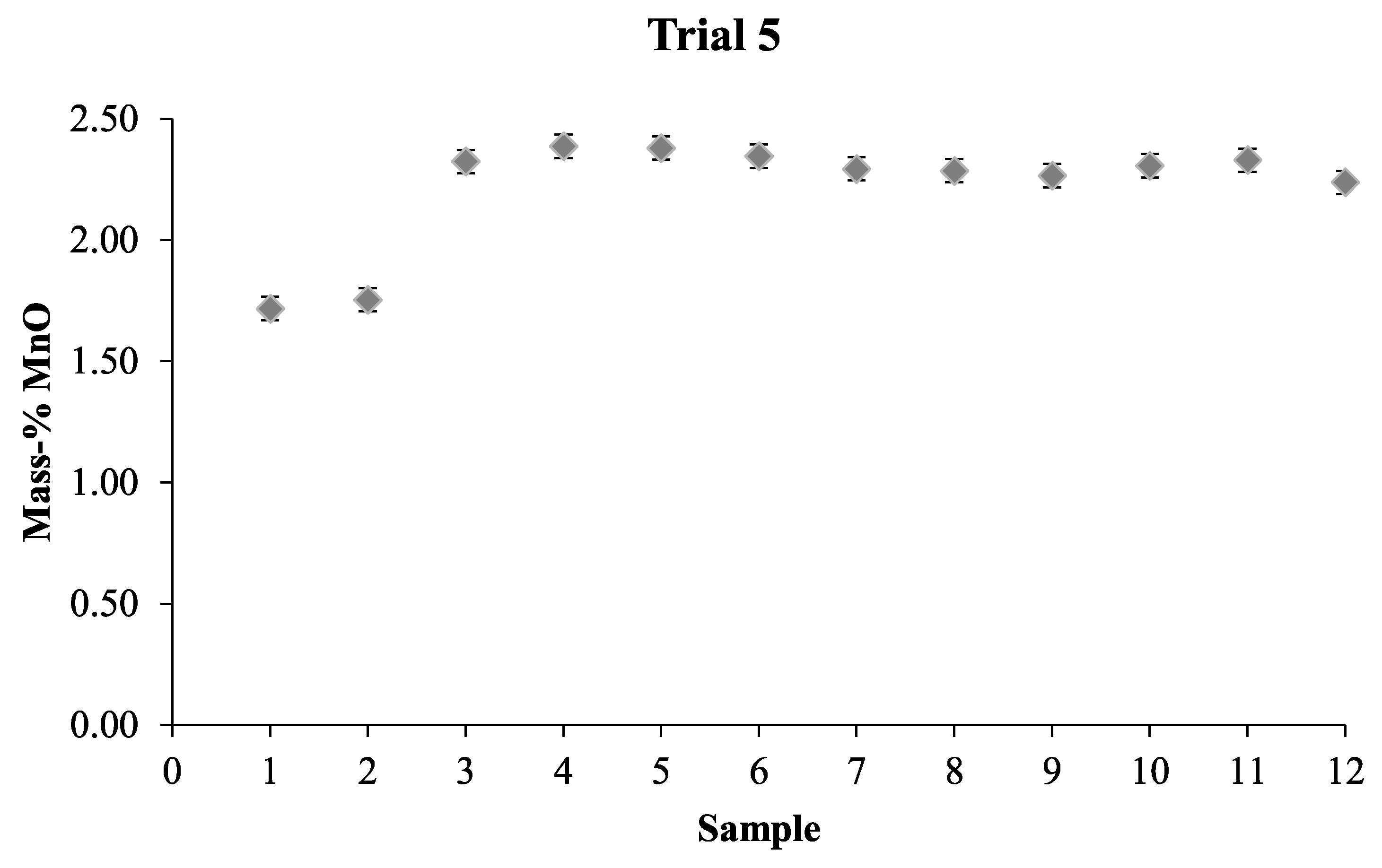
The composition of the slags used in all trials can be seen in
Table 8. The slag composition includes the element values that were used to normalize the data. In addition, Themo-Calc calculations were performed for all trials and for the respective slag. Only the elements representing a significant amount are shown in the table and these were also the amounts that were used as input data in the Thermo-Calc calculations. Specifically, elements larger than 1 mass-% were included in the Thermo-Calc calculations. This database uses 18 elements and is intended for solid or liquid sulfides or oxides and is used for slag calculations as well as for other applications [
21]. This was done in order to further investigate the phases of the slags in the different trials, in addition to the graphic LOM investigations that were made. Furthermore, it was used to investigate the amount of liquid phase in the slag during the operation of the process and to get a more in-depth knowledge to strengthen the earlier graphically presented LOM results. More specifically, the results indicate that a large majority of the slag was in a liquid state during the experiments.
As can be seen in
Table 8, the major slag elements were basically the same in each different trial. However, some differences can be seen between the slags. Namely, that the slag in Trials 1 and 2 had more than 54 mass-% FeO while the slag used in Trial 3 only contained 2.5 mass-% FeO. In the slag for Trial 4 there were small amounts of NiO present in the slag and more MnO was present in the slag in Trial 5 compared to the other trials. A low amount of MnO was present in all slags, since it was used as the tracer for the mixing time trials. Therefore, it was only considered for the slag used in Trial 5 when the amount of liquid phase was investigated using the Thermo-Calc calculations, since this slag had the highest amount of MnO.
The database TCOX7 was used for the calculations, as mentioned earlier. All slags with the compositions stated in
Table 8 were used. The operating slag temperatures were somewhere around 1200–1600 °C. For all Thermo-Calc calculations, both a closed system and an open system with an oxygen potential with the same value as the surrounding atmosphere were used. The oxygen potential was also varied for the different slags to be able to see the variation in the melting temperatures. The results from the calculations for the slags in Trials 1 and 2 are shown in
Figure 7,
Figure 8 and
Figure 9. These results were selected since they contained the highest amounts of FeO. Therefore, they are most important as the composition was closest to the composition of the future up-scaled IronArc production slag. These calculations were performed for atmospheric pressure. In
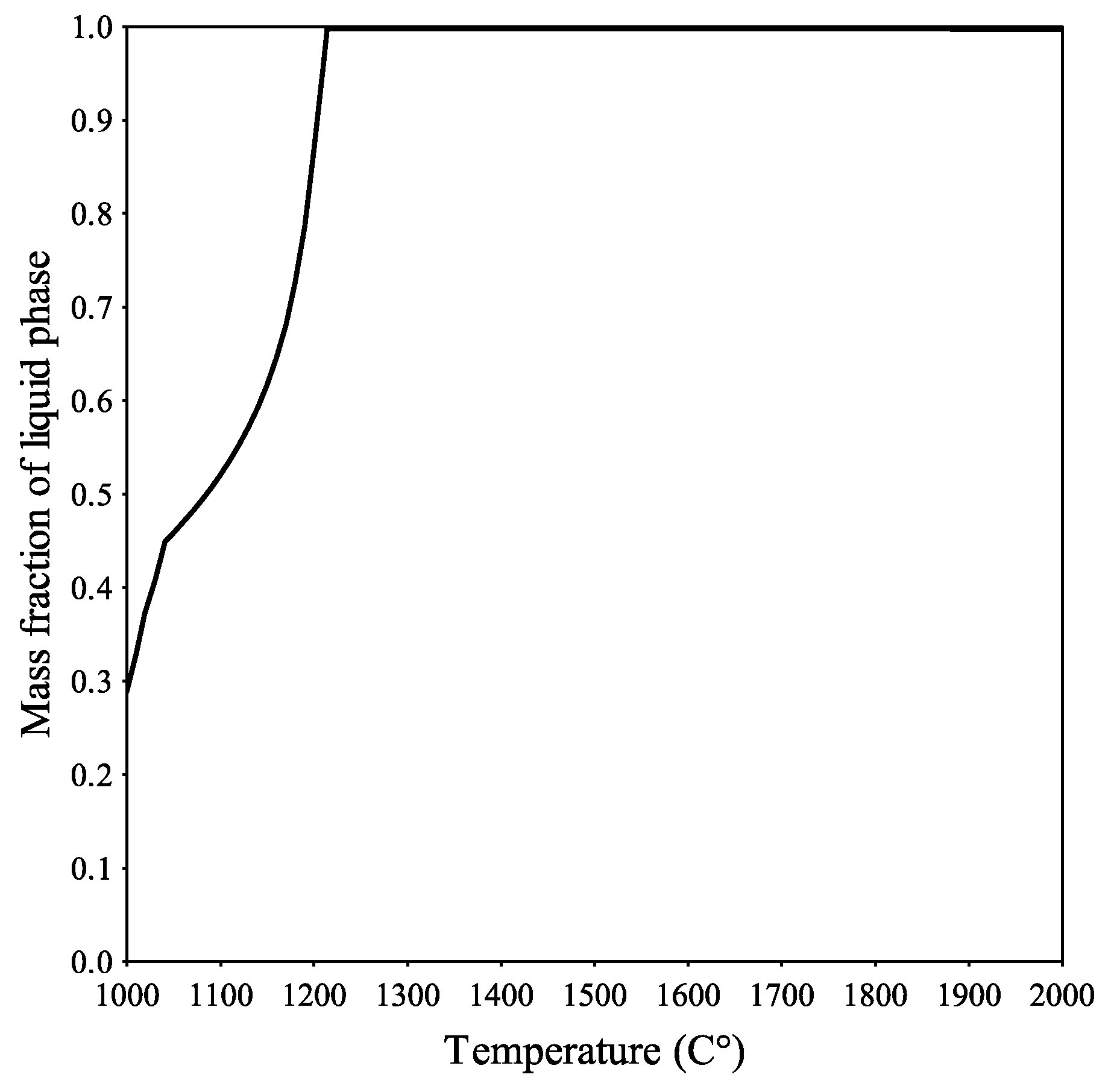
Figure 7, the results for the closed system are shown. According to the results obtained from the closed system, the slag at 1000 °C had a 30% liquid phase content and was completely melted and in a liquid state at 1250 °C. This is in line with the LOM investigation results that indicate that the slag was in a liquid state at the operating temperature.
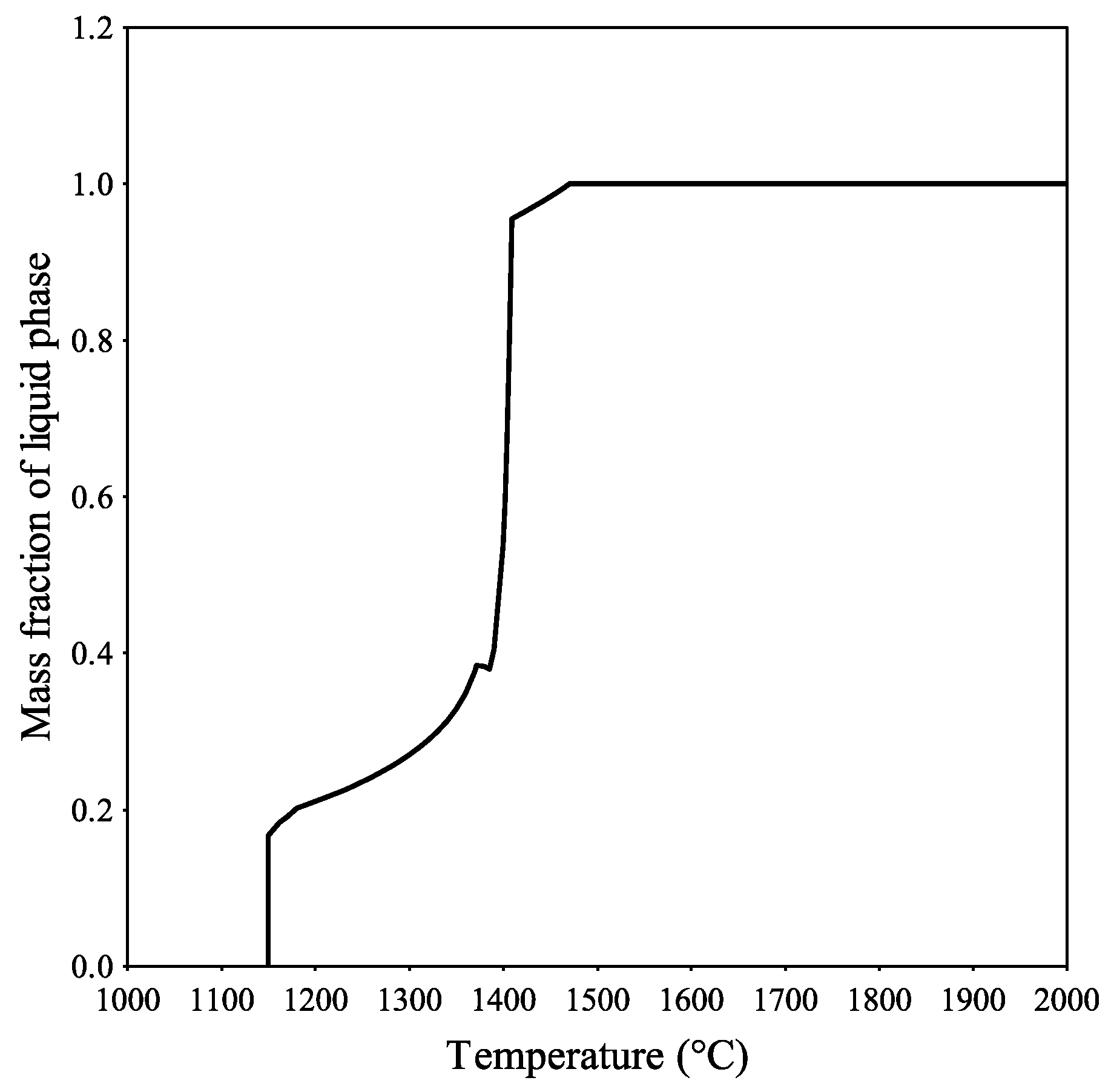
Figure 8 shows the mass fraction of the slag for an open system, where the slag is in equilibrium with an oxygen potential of 0.3.
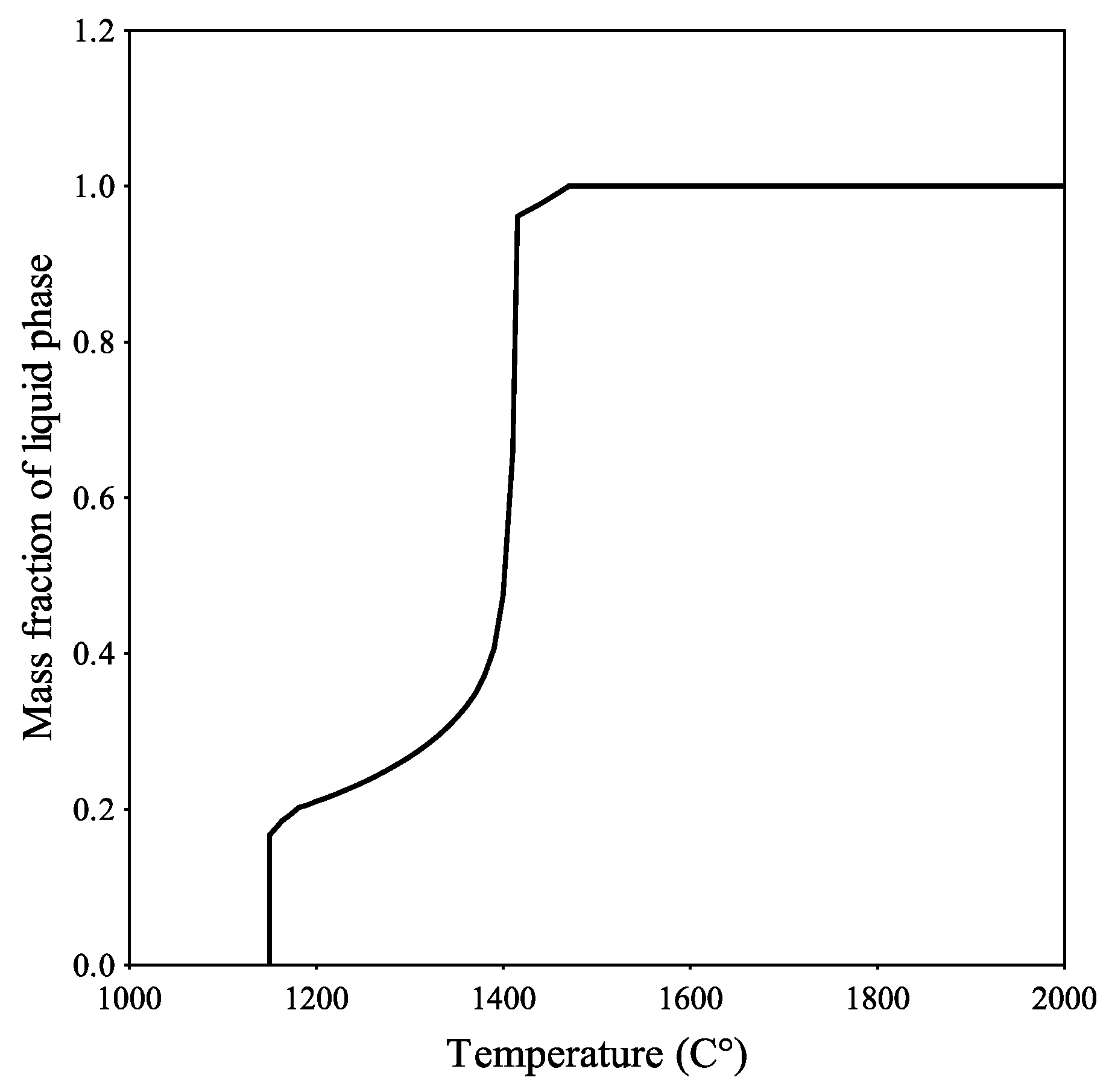
Figure 9 presents a similar calculation, but with an oxygen potential of 0.8. For both cases, the melting started at a higher temperature than given by the closed system and reached a liquid state higher than 90% at 1400 °C and were fully liquid at approximately 1470 °C. When observing the liquids line, the case seems to be that the oxygen potential did not affect the melting temperature to that extent, where the curves and a liquid amount of 95% differs by only 0.4%, when comparing the data for an oxygen potential value of 0.3 and 0.8 (
Figure 8 and
Figure 9). A completely liquid slag was reached at the same temperature. The larger difference was between an open and closed system, where the melting temperature was 1250 °C and 1450 °C according to the results for the slag elements in
Table 8. However, both the results obtained from an open and closed system indicate that the slag was in a liquid state during the operation of the pilot plant reactor. The results for the open system show a melting temperature that is closer to and on the limit of the pilot plant operation conditions. Similar calculations were done for Trials 3 to 5, for a closed system as well as for both higher and lower oxygen potentials. The results for these calculations were similar, but the closed system was closer in melting temperature to the open system compared to the slag used in Trials 1 and 2. In addition, the melting temperatures for these slags were predicted to be approximately 1400 °C. What should be noted is that the total number of elements in the slags is between 20 and 29, which is much higher than the number of elements that is used in the Thermo-Calc calculations. The amounts were reduced to ease the calculations and therefore only the elements with ≥1 mass-% were included. Since the number of elements in the actual slag was several times higher than in the calculations, the melting point should not have been higher than those in the results from the calculations. Often, a higher number of elements means a lower melting point for alloys compared to the pure metals of that alloy. Eutectic alloy systems often offer a lower melting point than the pure elements and also good fluidity [
22]. Furthermore, some of the elements that were not included in the calculations were sodium oxides, potassium oxides, and boron oxides. These elements work as fluxes and it is likely that the melting temperature of the slag would be even lower than in the Thermo-Calc calculations [
23,
24]. This is of interest since these elements are excluded in the Thermo-Calc calculations and therefore the melting point would be lower than the predicted calculation. Hence, the liquid slag conclusion is strengthened. Additionally, previous investigations have shown that for a slag containing Fe, Si, and O as the main elements, whereby they account for 90% of the slag, the main phase is fayalite and has a melting point of 1200 °C [
25]. The slags in Trials 1 and 2 also contain the main ion elements Fe, Si, and O, with a combined amount of 90.6 mass-%. This, in turn, gives further indications that the slag will be in a liquid state at the operating temperature of the pilot plant.
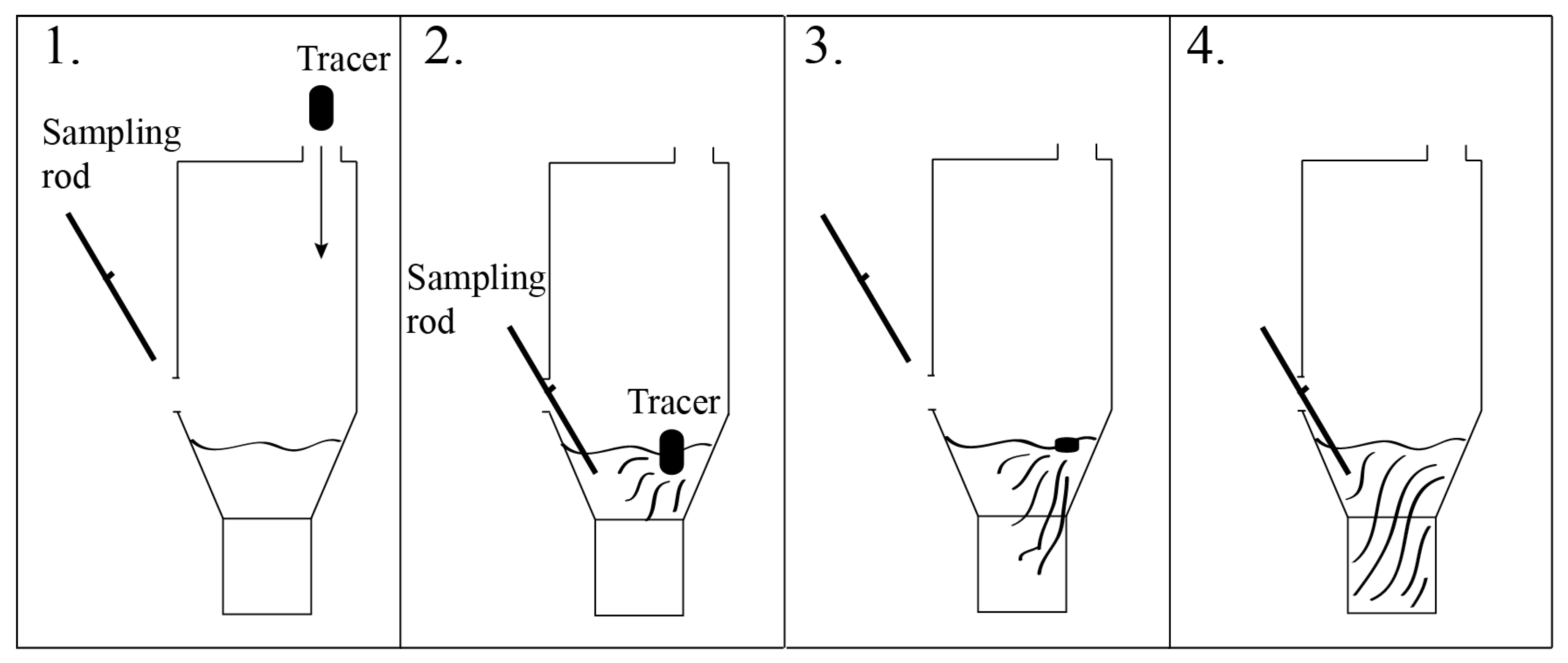
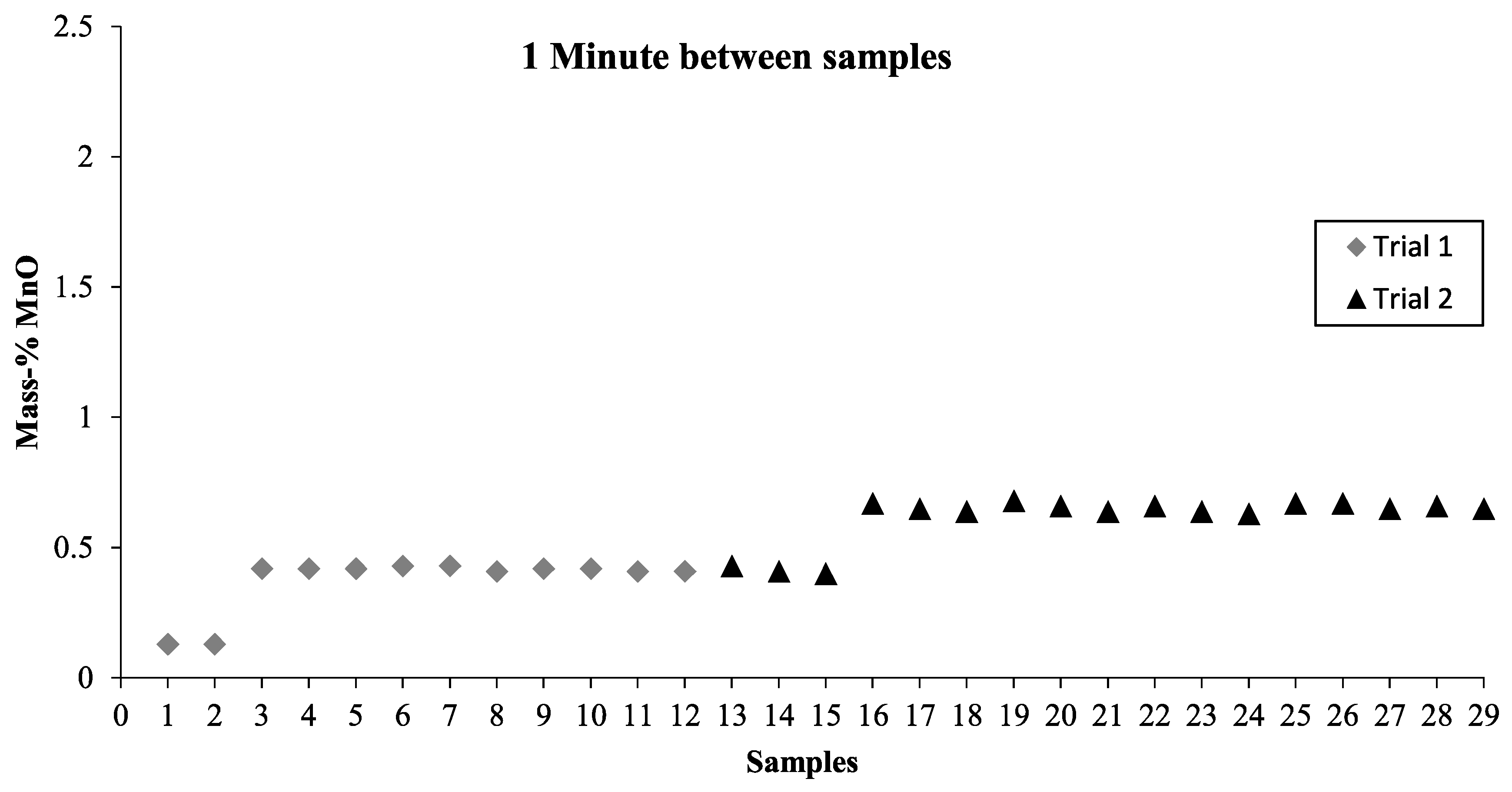
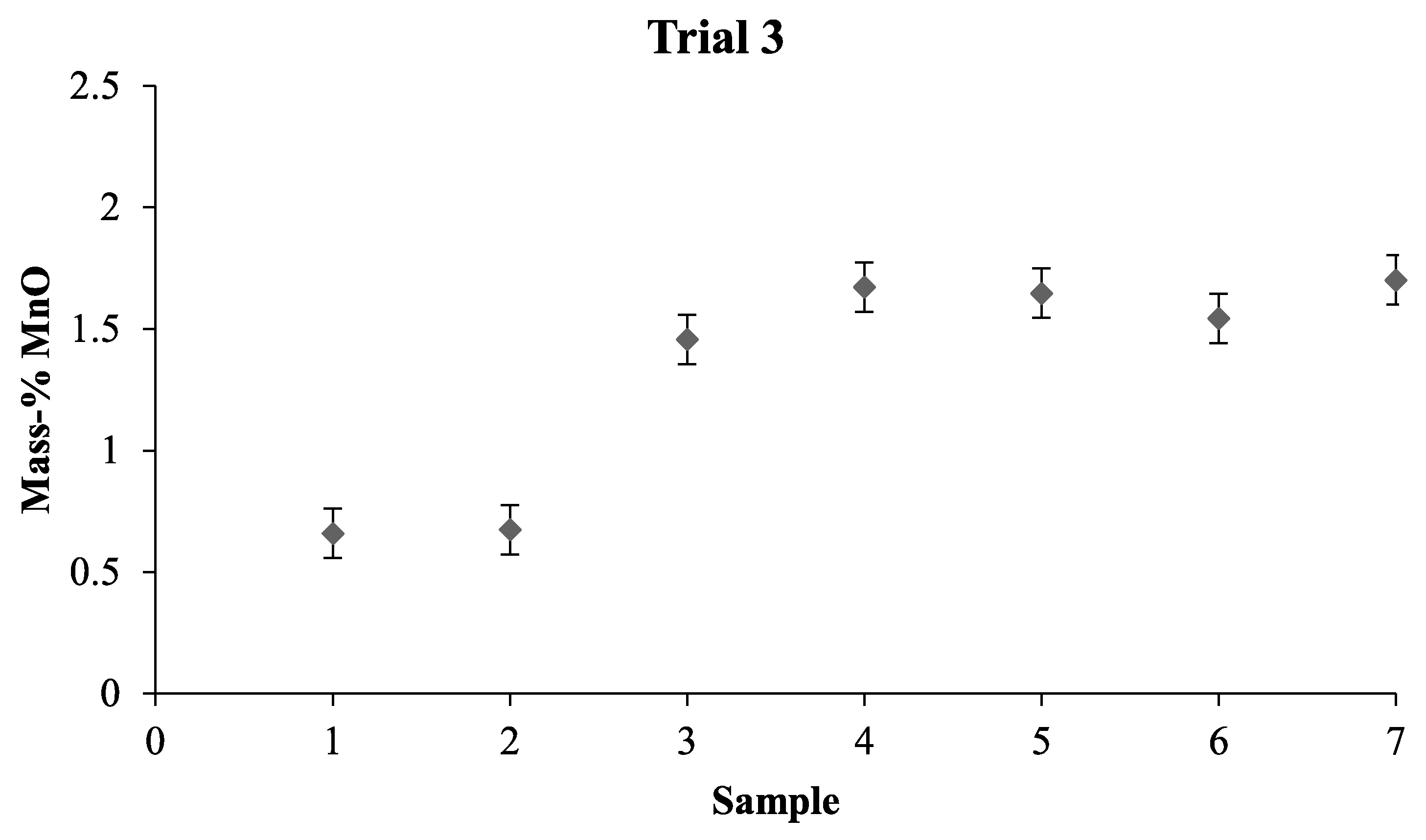
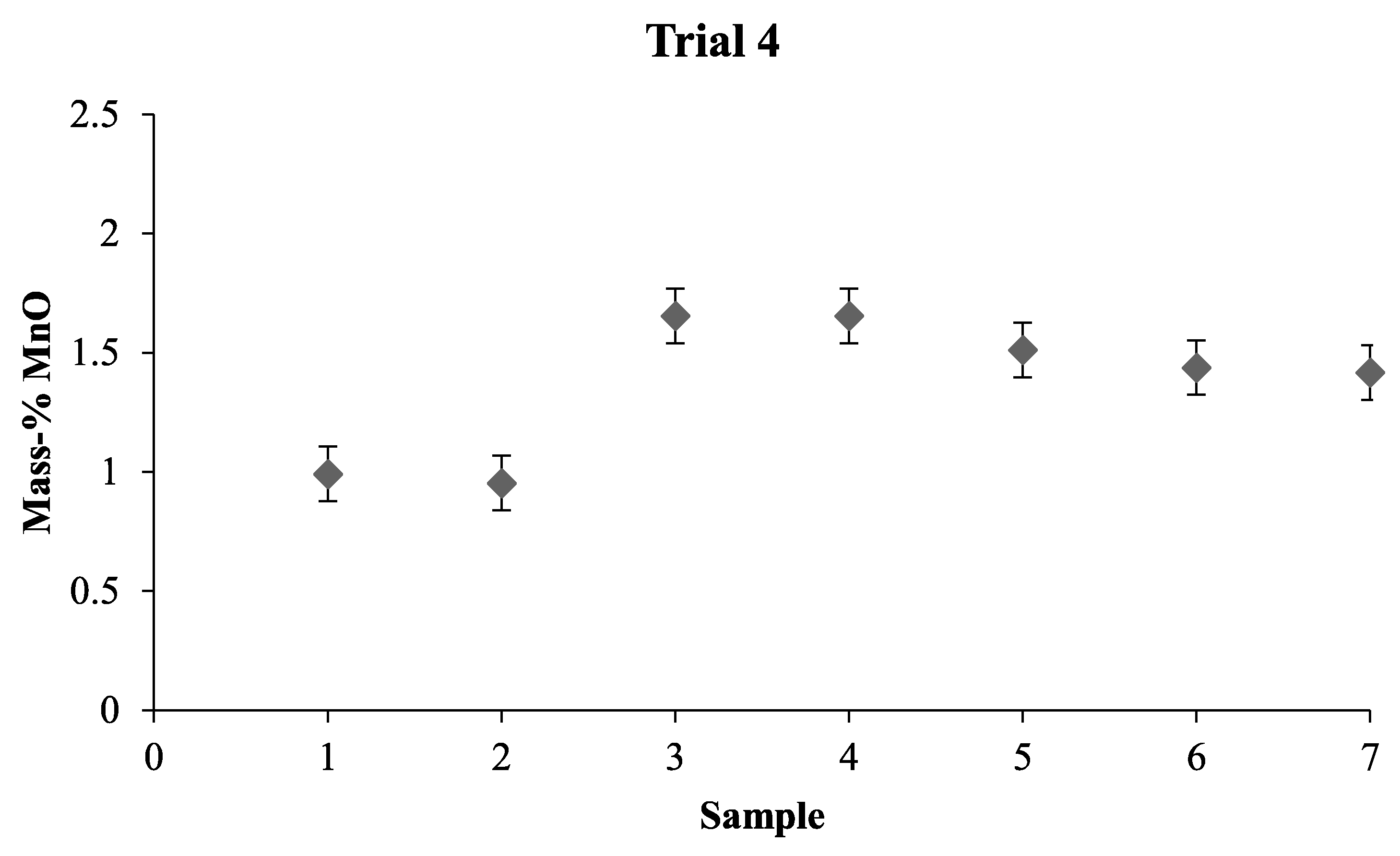
When combining the results from the LOM investigation, the mixing time experiments, and the Thermo-Calc calculations, we can assume that the slag in the pilot plant was in liquid state. For future mixing time investigations, it would be beneficial to take samples from different depths and different positions. This would give a clearer picture of the various parts of the bath. Furthermore, with the calculations made on the slag and the results obtained, numerical modeling can now be performed and used to predict the mixing time more accurately as a result of the knowledge that was gained from this investigation.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}