Abstract
Surface corrosion product formation is one of the important factors affecting the corrosion rate and hydrogen uptake in a H2S environment. However, it is still unclear how the base material composition will affect the corrosion products that are generated, and consequently their impact on the corrosion rate. In this paper, corrosion product formation and the impact of the Mo content of the base material on the composition of the corrosion products and hydrogen absorption in a sour environment was investigated. The corrosion layer was composed of a double layered mackinawite (FeS1−x) structure, which was enriched with molybdenum and chromium. The layers were formed via two different mechanisms, i.e., the inner layer was created via a general oxide film formation corrosion mechanism, whereas the upper layer was formed by a precipitation mechanism. The presence of this double corrosion layer had a large influence on the amount of diffusible hydrogen in the materials. This amount decreased as a function of contact time with the H2S saturated solution, while the corrosion rate of the materials shows no significant reduction. Therefore, the corrosion products are assumed to act as a physical barrier against hydrogen uptake. Mo addition caused a decrease in the maximal amount of diffusible hydrogen.
1. Introduction
In the oil and gas industry, the presence of H2S gas might induce sulfide stress cracking (SSC). The interaction between steel and the H2S containing surrounding environment also results in the formation of a FexSy corrosion product. Moreover, hydrogen is also generated during this corrosion process and can be absorbed into the material. Consequently, the potential threat for hydrogen induced damage is increased [1,2]. Furthermore, the characteristics of this FexSy surface layer will significantly affect the hydrogen introduction in the material [3,4].
Different FexSy phases can be formed when steel is in contact with an aqueous H2S containing environment. Shoesmith et al. [5] were amongst the first researchers in this field. They showed that the most prominent phase appeared to be mackinawite (tetragonal FeS1−x), which readily cracked and spalled from the metal surface. Other phases that were reported to be formed in H2S environments were cubic FeS, greigite, pyrrhotite, troilite, and pyrite [6]. The corrosion rate and relative amount of the different phases were strongly dependent on the environmental parameters, such as pH, applied current, and the degree of convection. Smith and Pacheco [7] determined the minimum amount of H2S required to produce mackinawite. Therefore, they generated a phase diagram showing the temperature and H2S range in which the different phases occur.
Nevertheless, the corrosion product layers have been determined to change over time [8]. Initially, the corrosion product consists of flake-like mackinawite, which grows larger and adheres with longer immersion time [9,10,11]. Consequently, an increase in the corrosion product film thickness is observed.
Multiple studies demonstrated that a non-homogeneous corrosion layer was found on the H2S exposed material and, moreover, the corrosion product films clearly consisted of different layers of iron sulfides [12]. Often, a two-layered corrosion film is observed, with the inner layer being adherent and dense, whereas the outer layer is more porous [11,13].
Generally, the corrosion resistance of a material is strongly dependent on its alloying elements. Molybdenum (Mo) is known to influence the corrosion resistance considerably [14]. Studies have shown that Mo addition to the base material enhanced the formation of the corrosion film, which hence, influenced the material’s corrosion behavior [15,16]. Molybdenum not only generated an improved the corrosion resistance, but it also formed carbides, which can trap diffusible hydrogen, as discussed in [13,17]. This type of hydrogen plays an important role in hydrogen induced failure [1]. Therefore, the presence of carbides is generally considered to be beneficial since they are assumed to trap hydrogen, efficiently removing detrimental diffusible hydrogen from the material.
A thorough understanding of the formation of the different corrosion products is of crucial importance since their presence could modify the kinetics of the corrosion process and hydrogen absorption inside the material [3,18]. The role of the corrosion products on the hydrogen/material interaction has been considered by hydrogen permeation in [3,18,19]. In some cases, it was demonstrated that the FeS corrosion products showed a protective nature since it can block the entry of H into the material [3,19]. However, this was not always the case, as this protective nature of the corrosion products was both temperature [3] and pH dependent [18]. These parameters have a large influence on the crystallographic composition of the FeS corrosion products. Other research demonstrated that the FeS is not protective and can even be considered as a reservoir of hydrogen nearby the metallic surface that can generate and sustain hydrogen absorption [20].
Surface corrosion product formation is one of the important factors affecting the corrosion rate and hydrogen uptake in a H2S environment. However, it is still unclear how the base material composition will affect the corrosion products that are generated, and consequently their impact on the corrosion rate. Moreover, the influence of the corrosion product on the hydrogen uptake is still under debate. In this study, two lab cast Quenched & Tempered (Q&T) steel, with different Mo content, were subjected to a sour environment for one month. The evolution of the corrosion products with time was characterized using scanning electron microscopy (SEM), electron probe micro-analyzer (EPMA), and X-ray diffraction (XRD). The amount of diffusible hydrogen was determined using hot extraction and the corrosion rate was evaluated by weight loss measurements. Consequently, the corrosion product formation and the impact of the Mo content of the base material on the composition of the corrosion products and hydrogen absorption with time in a sour environment was investigated.
2. Materials and Methods
The chemical composition of the two lab cast materials is presented in Table 1. Both materials have an identical composition, apart from the Mo content. Subsequent to casting, the materials were hot rolled to an overall thickness reduction of 84% in seven rolling passes. The materials were further austenitized for one hour at 890 °C to reach full austenitization and were quenched in water. The low Mo material was then tempered for one hour at two different temperatures, 670 and 700 °C, generating two different materials, i.e., low Mo (A) and low Mo (B), respectively. The high Mo material was tempered for one hour at 700 °C. A 0.2 offset yield strength (Rp0.2) of 638 MPa for the low Mo (A) material, 554 MPa for low Mo (B) and 706 MPa for the high Mo material was determined by performing tensile tests.

Table 1.
Compositions of the two lab cast materials.
The materials were cut in samples with dimensions of 30 mm length, 10 mm height, and 4 mm thickness. All the sides of the samples were ground and polished to 1 µm. Immersion tests were conducted at 25 °C in solution A of the NACE test TM0284 [21]. This consists of 5.0 wt % NaCl (VWR, Leuven, Belgium) and 0.5 wt % CH3COOH (VWR, Leuven, Belgium) in deionized water, produced by a Milli-Q Integral System, saturated with H2S from Air Liquide with a global purity >99.5% (max. impurities 500 ppm CH4, 3000 ppm Carbonyl Sulfide (COS)). Prior to saturation, the test solution was deaerated with N2 gas from Air Liquide (purity > 99.99%) for 40 min. After saturation and before the start of every test, the pH of the testing solution was measured to ensure that the value was below 2.8. The pH was measured after every test as well, to check if the value has not increased above 4. Samples were then removed from the vessel after 1, 2, 3, 4, 5, 7, 14, 21, and 28 days. This was performed via special in-house designed shaft, which allowed for the removal of samples without interrupting the test.
In order to study the morphology and to determine the chemical composition of the corrosion products, SEM images were taken of the surfaces and cross-sections of several samples, using a JSM-7600F FEG SEM of JEOL (JEOL, Akishima, Tokyo, Japan). Before the analysis of the cross-sections, the sample was ground and polished to 1 µm. To further characterize the features of the corrosion products in more detail, EPMA was performed, using a JXA-8530F set-up of JEOL (JEOL, Akishima, Tokyo, Japan). Additionally, the samples were analysed with XRD (Bruker, Karlsruhe, Germany) using a Siemens D5000 diffractometer with Mo Kα radiation to identify the formed corrosion products.
Hot extraction experiments were performed using a G8 Galileo with Thermal Conductivity Detector (TCD) from Bruker (Bruker, Karlsruhe, Germany) at 300 °C to determine the amount of diffusible hydrogen, according to the definition for diffusible hydrogen, as proposed by Wang et al. [22], and frequently applied since [23]. After extracting the samples from the vessel, they were stored in liquid nitrogen to minimise the loss of hydrogen. Prior to the hot extraction measurements, the samples were thoroughly ground, using a rotary tool, to remove all of the corrosion products and rinsed with acetone.
The materials were also electrochemically charged to determine the hydrogen content. This cathodic charging was performed for one hour, using a 0.5 M sulfuric acid aqueous solution containing 1 g/L thiourea at a current density of 0.8 mA/cm². After charging, the materials were rinsed with water and acetone, dried, and the hydrogen content was measured using hot extraction, as described above.
Weight loss measurements were performed using the ASTM G1-90 solution, also known as the Clarke solution [24]. This consists of concentrated HCl with 2 wt % Sb2O3 and 5 wt % SnCl2. The samples were scrubbed before they were put in the solution for 30 min, while vigorously stirring, and rinsed with acetone.
3. Results and Discussion
3.1. Corrosion Product Characterization
3.1.1. Surface Topology and X-ray Diffraction (XRD) Measurements
The evolution of the corrosion products formed on the surface of the materials was investigated with SEM. Top view images are presented in Figure 1, where only the SEM images of the low Mo (A) and high Mo materials are shown, as all of the materials gave very similar results. After three days, only very small nanocrystals were observed on the surface, whereas 14 days contact time clearly resulted in crystal growth of the corrosion products on the surface of all alloys. These crystals grow further as a function of test duration, as seen after 28 days. Similar crystal structures were observed by Bai et al. [25] in their corrosion products investigation on a carbon steel in a hydrogen sulfide environment.

Figure 1.
Top view images of the low Mo (A) material after (a) 3 days, (b) 14 days, and (c) 28 days and of the high Molybdenum (Mo) material after (d) 3 days, (e) 14 days, and (f) 28 days.
XRD measurements allowed further identification of the corrosion products. In Figure 2, XRD patterns of the corrosion products that were formed after 14 and 28 days in the test solution on the low Mo (A) and high Mo materials are shown, as well as the XRD pattern of the corrosion product residue that was recovered from the bottom of the cell after the test. On the surfaces of both materials, similar corrosion products were found, namely mackinawite (FeS1−x), which was in agreement with the general assumption that mackinawite was the first formed corrosion product in an H2S environment under ambient conditions [5,25,26]. The increasing intensity of the major diffraction peak of mackinawite, the {001} reflection, as a function of the test duration, indicated that a larger degree of mackinawite is formed in a preferential orientation. Additionally, peak intensities were further increasing with longer test durations, which indicated an increase of the amount of formed mackinawite. When comparing the diffraction spectra of the corrosion products after 14 days and 28 days, a peak shift of the {001} diffraction peak by ±0.1° to higher diffraction angles was observed, see Figure 2a–c. This corresponds to a lattice contraction along the c-axis, which generated additional strain in the corrosion products, and as such, this will contribute to the poor adhesion of the corrosion products to the substrate. This was in good agreement with the observation by Genchev et al. [11].
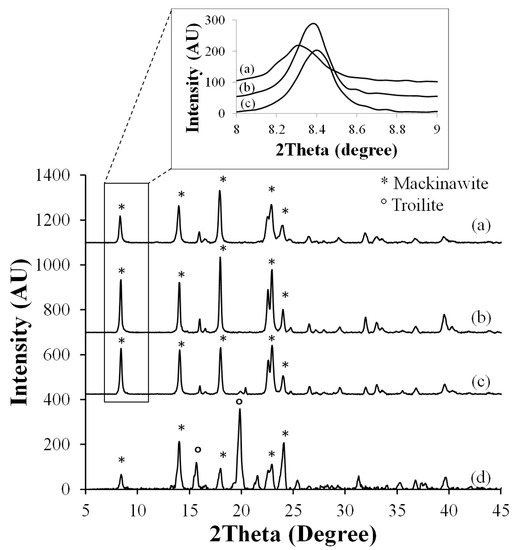
Figure 2.
X-ray diffraction (XRD) patterns of corrosion products formed (a) after 14 days on the low Mo (A) material, (b) after 28 days on the low Mo (A) material, (c) after 28 days on the high Mo material, and (d) the corrosion product residue that was found on the bottom of the cell after testing. And an enlargement of the first peak of patterns a-c (inset).
The XRD pattern of the corrosion product residue, which was collected after the test and consisted out of corrosion products that spalled of the materials during the entire test, was composed of mackinawite and troilite (hexagonal FeS). According to Bai et al. [25], troilite is generated outside the iron-rich mackinawite, because the unbroken mackinawite layer prevents the diffusion of iron ions towards the solid-liquid interface. Since the troilite was generated at the outer surface of the material, this phase will easily be removed from the sample surface during the test and end up as residue at the bottom of the cell.
3.1.2. Cross-Section Analysis
A two-layered structure was clearly visible when investigating the cross-sections of the corrosion products in Figure 3. The upper layer appeared to be brittle and was very often missing. The corrosion product underneath was more adherent to the surface of the material. Since the outer layer had a very variable thickness, only the thickness of the inner layer was used to evaluate the corrosion product thickness. The inner layer grew to reach a value after 28 days of 54 ± 15, 46 ± 16, and 73 ± 18 µm for the low Mo (A), low Mo (B), and high Mo material, respectively. The presence of a double layer indicated that two mechanisms are generating the corrosion products. Initially, the first layer of mackinawite was formed via a solid-state reaction [4,27]:
Fe(s) + (1 − x)H2S → FeS1−x(s) + (1 − x)H2,

Figure 3.
Scanning electron microscopy (SEM) images of the cross-sections of the low Mo (A) material (left), low Mo (B) material (middle), and high Mo material (right).
Therefore, the inner layer was generated by a uniform corrosion process. After this first step of corrosion product formation, sulfides were also generated by a precipitation mechanism [27], which created the upper layer. Mackinawite formation was much faster than the formation of other thermodynamically stable products, such as troilite and pyrrhotite [4]. The precipitation occurred according to the following reaction scheme:
Fe + H2S + H2O → FeSH−ads + H3O+,
Fe + HS− → FeSH−ads,
FeSH−ads ⇔ FeSH+ads + 2e−,
The adsorbed FeSH+ complex can be directly incorporated in the mackinawite layer or it can undergo hydrolysis to generate Fe2+, as shown in the reactions below:
FeSH+ads → FeS1−x (s) + xSH− + (1 − x)H+,
FeSH+ads + H3O+ → Fe2+ + H2S + H2O,
The elemental distribution in the corrosion product layers was visualised by performing EPMA measurements. Figure 4, Figure 5 and Figure 6 show the Backscattered Electron (BSE) images and elemental mappings of the three materials. The two-layered structure of the corrosion products on all of the materials was clearly demonstrated as both layers clearly have a different composition. The upper layer consisted mainly of Fe and S enriched with Mo, while the inner layer consisted of Fe and S enriched with Cr and Mo.
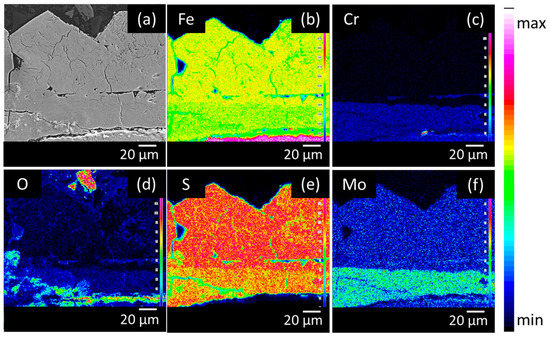
Figure 4.
Cross-sectional Backscattered Electron (BSE) image (a) and elemental mappings of (b) Fe, (c) Cr, (d) O, (e) S and (f) Mo of the low Mo (A) material after 28 days of immersion in the H2S saturated test solution.
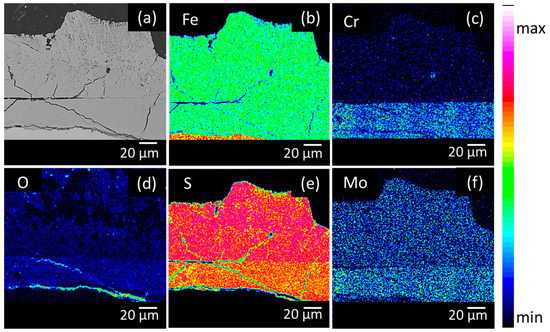
Figure 5.
Cross-sectional BSE image (a) and elemental mappings of (b) Fe, (c) Cr, (d) O, (e) S and (f) Mo of the low Mo (B) material after 28 days of immersion in the H2S saturated test solution.
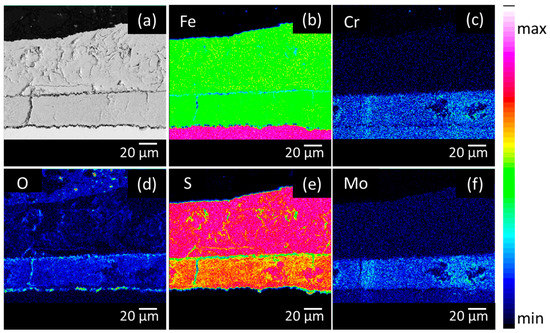
Figure 6.
Cross-sectional BSE image (a) and elemental mappings of (b) Fe, (c) Cr, (d) O, (e) S and (f) Mo of the high Mo material after 28 days of immersion in the H2S saturated test solution.
The quantitative elemental compositions that were obtained from the EPMA measurements are shown in Table 2. The high amount of both Fe and S for the upper layer confirmed the XRD results, which showed that the upper layer was composed of mackinawite. Additionally, a similar amount of Mo was present in the upper layer of all materials, which corresponded with the assumption that the upper layer was created via a precipitation mechanism. Since Mo has a high affinity for S, MoS2 species will be generated and precipitate together with the FeS1−x. Cr, on the other hand, was not present in the upper layer. These findings were in correspondence with the equilibrium phase diagrams of both Mo and Cr in Figure 7. These equilibrium phase diagrams were made with HSC Chemistry, using an estimation of the concentrations of the different elements that were present at a specific place in the solution. Two different situations were simulated, Figure 7A,C show the situation near the surface of the metal, where the S concentration is lower (1 × 10−3 M) and the Fe concentration is higher (1 M), while Figure 7B,D show the situation of the corrosion product in contact with the electrolyte Figure 7B confirms the formation of MoS2 as corrosion product that was above mentioned to be generated by a precipitation mechanism. Figure 7D shows that this is not the case for Cr, which will dissolve into the solution as Cr3+ and will not precipitate. This confirms the EPMA results, showing no Cr in the upper layer of the corrosion products.
Table 2.
Mean compositions of the upper and inner corrosion layer of the low Mo (A), low Mo (B), and high Mo materials, measured with electron probe micro-analyzer (EPMA).
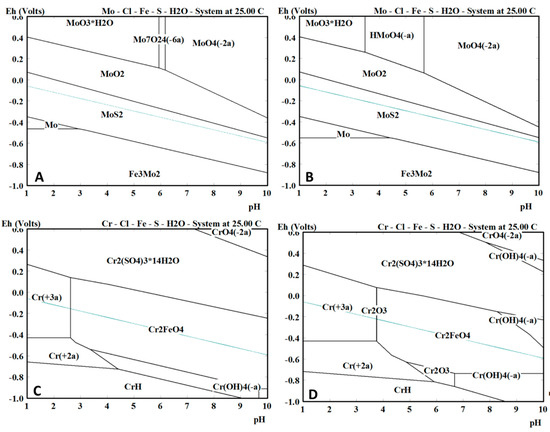
Figure 7.
The equilibrium phase diagram obtained using HSC Chemistry for the Mo and Cr specimens near the surface (A,C, respectively) and further away from the surface (B,D, respectively). During the test a pH value between 2.8 and 4.0 was maintained.
The inner layer was mainly composed of Fe, S, Mo, and Cr. For both of the materials, the inner layers of the corrosion products were enriched with Mo in comparison to the base materials. A similar observation was made for stainless steels during active dissolution, the corrosion products had a higher Mo content than the bulk material [28]. The Mo amount in the inner layer was remarkably different. The amount of Mo in the inner layer of the high Mo material was more than twice that of the Mo in the inner layer of both low Mo materials.
Chromium was only present in the inner layer of the corrosion products and in the base material. Cr will react selectively with O [14,29], the increased amount of O (cf. Figure 4, Figure 5 and Figure 6) in the inner layer confirmed this assumption. Figure 7 again supported these results. Figure 7A shows the formation of MoS2 near the surface. Figure 7C shows that closer to the surface, in the presence of a higher concentration of Fe, Cr2FeO4 will be formed. On the EPMA images (cf. Figure 4, Figure 5 and Figure 6) a highly elevated concentration of O is visible in a thin layer above the metal surface. This could be a Cr2FeO4 layer that was formed. The formation of an oxide layer in between the mackinawite and the base metal was also observed by Genchev et al. [11]. This oxide layer could cause a deterioration of the adhesion of the corrosion products to the surface, and as such, have an influence on the hydrogen uptake and the corrosion rate [30].
3.2. Hydrogen Uptake and Corrosion Rate Evaluation
In order to evaluate the effect of the corrosion layer on the hydrogen uptake, the amount of diffusible hydrogen present in the samples was measured as a function of the test duration and is summarized in Figure 8. A peak in the amount of diffusible hydrogen was clearly observed for all materials. However, for the low Mo (A) material the amount of diffusible hydrogen was much higher, especially after submersion times of two weeks and less. This could be related to the amount of dislocations that are present in the material, which have been demonstrated to trap hydrogen with low activation energy [31]. Since the low Mo (A) material was tempered at a lower temperature, cf. the experimental procedure, the amount of dislocations will be higher, causing the amount of maximal diffusible hydrogen to be higher. To confirm this statement, the base materials were also charged electrochemically, without immersion in H2S, and the amount of diffusible hydrogen was determined, as can be seen in the inset of Figure 8. Since a similar trend was found in amount of diffusible hydrogen after electrochemical charging, it could be concluded that the difference in peak values was intrinsic to the material and related to their dislocation density.
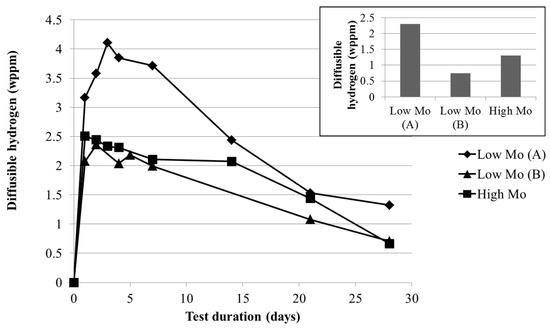
Figure 8.
Amount of diffusible hydrogen, determined using a hot extraction technique, present in the low Mo (A), the low Mo (B), and the high Mo material as a function of the test duration and after electrochemical charging (inset).
After the initial peak, the amount of diffusible hydrogen was decreasing rapidly for all of the materials. This decline could have several origins. On the one hand, cracks could be generated in the materials through Hydrogen Induced Cracking (HIC), rendering the formation of H2 in the cracks and a reduction in the amount of diffusible hydrogen. Therefore, all materials were investigated by cross-sectional analysis in order to detect the presence of cracks. Only in the low Mo (A) material that was in the test solution of 28 days, a crack was observed, as can be seen in Figure 9. However, since this HIC crack only occurred at the end of the testing period, it not likely to be the cause of the decline of the amount of diffusible hydrogen, since this already started after 1 to 3 days of testing. On the other hand, the decline of diffusible hydrogen could also be caused by a diminishing generation of hydrogen. Hence, the corrosion rate as a function of test duration was investigated.

Figure 9.
Optical microscope image of a hydrogen induced crack present in the low Mo material after 28 days of testing.
The corrosion rate (CR) evolution during the test was followed by performing weight loss measurements, the corrosion rate as a function of the test duration can be seen in Figure 10. The CR does not decline as a function of time. This implies that the corrosion products that are generated during the test are not protective against further corrosion, in spite of the constant decrease of the amount of diffusible hydrogen in the material.
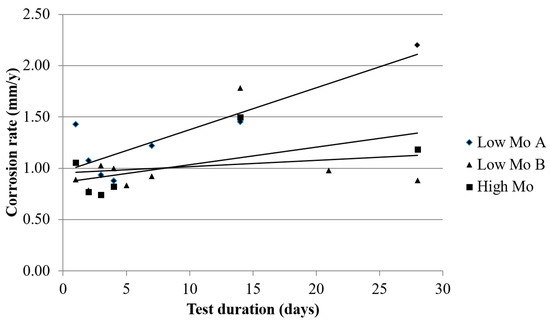
Figure 10.
The corrosion rate of the low Mo (A), the low Mo (B), and the high Mo materials as a function of the test duration.
A general schematic overview of the corrosion product generation and its influence on the diffusible hydrogen content is shown in Figure 11. The first generation of FeS corrosion product caused a first steep increase of the amount of diffusible hydrogen in the steel. After a few days, a double layer of FeS corrosion products was formed. The outer layer was generated via a deposition mechanism and was very often missing, indicating its poor adhesion. The inner layer could be formed by the diffusion of Fe from the metal through the FeS layer or by the diffusion of the S containing species through the FeS layer to the metal. The latter is assumed to occur, since the original flat surface of the sample was still clearly visible in the corrosion products, as can be observed in Figure 3. In addition, the FeS layer growth could be induced by H2S solid state diffusion through the crystalline layer or by ionic conduction of S2− and HS−. Both of the situations would imply a decrease in the amount of diffusible hydrogen, as was experimentally observed. On the one hand, the H2S solid state diffusion could create a decrease in the diffusible hydrogen content that is caused by a decrease in the corrosion rate. On the other hand, during the ionic conduction of S2− and HS−, a lot of hydrogen is already released outside the FeS layer and will be lost in the environment as H2. Since the corrosion rate does not clearly decrease as a function of time, as can be seen in Figure 10, the ionic conduction of S2− and HS− is believed to be the mechanism of FeS formation during these tests. These results are in agreement with the results of Huang et al. [19]. Using hydrogen permeation, they observed a decrease in the steady-state hydrogen permeation current and the hydrogen diffusivity. This was attributed to the corrosion product film being an n-type semiconductor with an anion selectivity. As such, S2− and HS− ions are attracted to and H+ ions are repealed from surface.
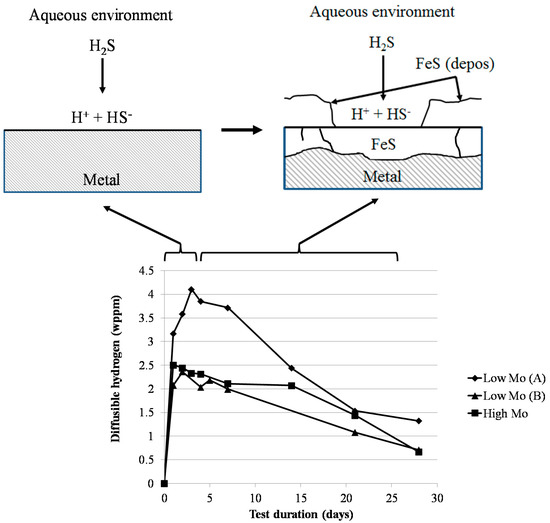
Figure 11.
Schematic overview of the corrosion product formation and the influence of this corrosion product on the hydrogen content inside the metal.
4. Conclusions
In this paper, corrosion product formation and the impact of the Mo content of the base material on the composition of the corrosion products and hydrogen absorption with time in a sour environment was investigated.
The corrosion products that were generated in an aqueous solution saturated with H2S had a two-layered structure. The main composition of both layers was mackinawite (FeS1−x), however, they contained different amounts of alloying elements, molybdenum, and chromium. The upper layer, which was created via a precipitation mechanism, was enriched with Mo. While the inner layer was formed via a uniform corrosion process and was enriched with Mo and Cr. Investigation of the equilibrium phase diagrams for these environments showed that indeed Mo can undergo a deposition reaction, creating MoS2, while Cr will not undergo any deposition, but remain in solution as Cr2+.
For all three of the materials that were tested, a similar trend in diffusible hydrogen content over time was observed. The amount of diffusible hydrogen reached a peak in the beginning of the test and declined after this first peak. The maximum amount of diffusible hydrogen was much higher for the low Mo (A). This was attributed to the higher amount of dislocations present in the material. The decline of the amount of diffusible hydrogen is believed to be due to the presence of the corrosion products. Since the ionic conduction of S2− and HS− is most likely the formation mechanism of these corrosion products, a lot of hydrogen is already released outside the FeS layer and will be lost in the environment as H2. As such, the corrosion product formation can help to prevent hydrogen uptake in the material.
The difference in Mo of the two different materials did not seem to have any effect on the corrosion rate or the hydrogen uptake in H2S environment. The biggest advantage of Mo addition to the base material in this research is the higher temper softening resistance that is caused by the higher amount of Mo. This allows for the material to be tempered for a longer amount of time without major loss of strength, causing the dislocation density to be lower and the maximal amount of diffusible hydrogen to decline.
Acknowledgments
The authors wish to thank the Special Research Fund (BOF) (BOF15/BAS/062 and 01P03516) and Ghent University. The authors also acknowledge the technicians and staff working at the sour laboratory at OCAS (ArcelorMittal R&D Gent) and the technical staff from the Department of Materials, Textiles and Chemical Engineering, Ghent University, for their help with the experiments and sample preparation.
Author Contributions
Elien Wallaert and Kim Verbeken designed the experiments, Elien Wallaert conceived and performed the experiments; Elien Wallaert, Tom Depover and Kim Verbeken analyzed the data; Kim Verbeken and Iris De Graeve contributed analysis tools; Elien Wallaert wrote the paper. The other authors provided feedback on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Depover, T.; Wallaert, E.; Verbeken, K. Fractographic analysis of the role of hydrogen diffusion on the hydrogen embrittlement susceptibility of dp steel. Mater. Sci. Eng. A 2016, 649, 201–208. [Google Scholar] [CrossRef]
- Depover, T.; Escobar, D.P.; Wallaert, E.; Zermout, Z.; Verbeken, K. Effect of hydrogen charging on the mechanical properties of advanced high strength steels. Int. J. Hydrogen Energy 2014, 39, 4647–4656. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Zhou, C.S.; Wang, P.Y.; Chen, C.F.; Chen, L.Q. Effects of the temperature on the hydrogen permeation behaviours of l360NCS pipeline steel in 1 MPa H2S environments. Int. J. Electrochem. Sci. 2013, 8, 2880–2891. [Google Scholar]
- Zhou, C.S.; Chen, X.Y.; Wang, Z.; Zheng, S.Q.; Li, X.; Zhang, L. Effects of environmental conditions on hydrogen permeation of X52 pipeline steel exposed to high H2S-containing solutions. Corros. Sci. 2014, 89, 30–37. [Google Scholar] [CrossRef]
- Shoesmith, D.W.; Taylor, P.; Bailey, M.G.; Owen, D.G. The formation of ferrous monosulfide polymorphs during the corrosion of iron by aqueous hydrogen sulfide at 21 °C. J. Electrochem. Soc. 1980, 127, 1007–1015. [Google Scholar] [CrossRef]
- Smith, S.N.; Joosten, M.W. Corrosion of Carbon Steel by H2S in CO2 Containing Oilfield Environments; Corrosion 2006, Paper No. 06115; NACE International: Houston, TX, USA, 2006. [Google Scholar]
- Smith, S.N.; Pacheco, J.L. Prediction of Corrosion in Slightly Sour Environments; Corrosion 2003, Paper No. 02241; NACE International: Houston, TX, USA, 2002. [Google Scholar]
- Sosa, E.; Cabrera-Sierra, R.; Oropeza, M.T.; Hernandez, F.; Casillas, N.; Tremont, R.; Cabrera, C.; Gonzalez, I. Chemical characterization of corrosion films electrochemically grown on carbon steel in alkaline sour environment. J. Electrochem. Soc. 2003, 150, B530–B535. [Google Scholar] [CrossRef]
- Bai, P.P.; Zheng, S.Q.; Chen, C.F. Electrochemical characteristics of the early corrosion stages of api X52 steel exposed to H2S environments. Mater. Chem. Phys. 2015, 149, 295–301. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Ke, W.; Han, E.-H. Corrosion behavior of X52 anti-H2S pipeline steel exposed to high H2S concentration solutions at 90 degrees C. J. Mater. Sci. Technol. 2014, 30, 504–510. [Google Scholar] [CrossRef]
- Genchev, G.; Cox, K.; The Hai, T.; Sarfraz, A.; Bosch, C.; Spiegel, M.; Erbe, A. Metallic, oxygen-containing reaction products after polarisation of iron in H2S saturated saline solutions. Corros. Sci. 2015, 98, 725–736. [Google Scholar] [CrossRef]
- Kvarekval, J.N.R. Formation of Multilayer Iron Sulfide Films during High Temperature Co2/H2s Corrosion of Carbon Steel; Corrosion 2003, Paper No. 03339; NACE International: Houston, TX, USA, 2003. [Google Scholar]
- Craig, B.D. The role of Mo in the enhanced resistance to hydrogen stress cracking of ALSL-4100 steels. Metall. Trans. A-Phys. Metall. Mater. Sci. 1982, 13, 1099–1101. [Google Scholar] [CrossRef]
- Marcus, P. Sulfur-assisted corrosion mechanisms and the role of alloyed elements. In Corrosion Mechanisms in Theory and Practice, 2nd ed.; Marcus, P., Ed.; CRC Press: Boca Raton, FL, USA, 2002; pp. 404–406. [Google Scholar]
- Genchev, G.; Bosch, C.; Wanzenberg, E.; Erbe, A. Role of molybdenum in corrosion of iron-based alloys in contact with hydrogen sulfide containing solution. Mater. Corros. 2017, 68, 595–603. [Google Scholar] [CrossRef]
- Monnot, M.; Roche, V.; Estevez, R.; Mantel, M.; Nogueira, R.P. Molybdenum effect on the sulfide stress corrosion of a super martensitic stainless steel in sour environment highlighted by electrochemical impedance spectroscopy. Electrochim. Acta 2017, 252, 58–66. [Google Scholar] [CrossRef]
- Depover, T.; Verbeken, K. Evaluation of the role of Mo2C in hydrogen induced ductility loss in Q&T Fe-C-Mo alloys. Int. J. Hydrogrn Energy 2016, 41, 14310–14329. [Google Scholar]
- Zhou, C.S.; Zheng, S.Q.; Chen, C.F.; Lu, G.W. The effect of the partial pressure of H2S on the permeation of hydrogen in low carbon pipeline steel. Corros. Sci. 2013, 67, 184–192. [Google Scholar] [CrossRef]
- Huang, F.; Cheng, P.; Zhao, X.Y.; Liu, J.; Hu, Q.; Cheng, Y.F. Effect of sulfide films formed on X65 steel surface on hydrogen permeation in H2S environments. Int. J. Hydrogen Energy 2017, 42, 4561–4570. [Google Scholar] [CrossRef]
- Monnot, M.; Nogueira, R.P.; Roche, V.; Berthome, G.; Chauveau, E.; Estevez, R.; Mantel, M. Sulfide stress corrosion study of a super martensitic stainless steel in H2S sour environments: Metallic sulfides formation and hydrogen embrittlement. Appl. Surf. Sci. 2017, 394, 132–141. [Google Scholar] [CrossRef]
- NACE International, Nace standard TM0284-2003. In NACE Standard TM0284-2003, Evaluation of Pipeline and Pressure Vessel Steels for Resistance to Hydrogen-Induced Cracking; NACE International: Houston, TX, USA, 2003.
- Wang, M.; Akiyama, E.; Tsuzaki, K. Effect of hydrogen on the fracture behavior of high strength steel during slow strain rate test. Corros. Sci. 2007, 49, 4081–4097. [Google Scholar] [CrossRef]
- Pérez Escobar, D.; Wallaert, E.; Duprez, L.; Atrens, A.; Verbeken, K. Thermal desorption spectroscopy study of the interaction of hydrogen with tic precipitates. Met. Mater. Int. 2013, 19, 741–748. [Google Scholar] [CrossRef]
- ASTM International. G 1–90 Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens; ASTM International: West Conshohocken, PA, USA, 1992. [Google Scholar]
- Bai, P.; Zheng, S.; Zhao, H.; Ding, Y.; Wu, J.; Chen, C. Investigations of the diverse corrosion products on steel in a hydrogen sulfide environment. Corros. Sci. 2014, 87, 397–406. [Google Scholar] [CrossRef]
- Qi, Y.; Luo, H.; Zheng, S.; Chen, C.; Wang, D. Effect of immersion time on the hydrogen content and tensile properties of A350LF2 steel exposed to hydrogen sulphide environments. Corros. Sci. 2013, 69, 164–174. [Google Scholar] [CrossRef]
- Sun, W. Kinetics of Iron Carbonate and Iron Sulfide Scale Formation in CO2/H2S Corrosion. Ph.D. Thesis, Ohio University, Athens, OH, USA, 2006. [Google Scholar]
- Qvarfort, R. Some observations regarding the influence of molybdenum on the pitting corrosion resistance of stainless steels. Corros. Sci. 1998, 40, 215–223. [Google Scholar] [CrossRef]
- Cheng, X.L.; Ma, H.Y.; Chen, S.H.; Niu, L.; Lei, S.B.; Yu, R.; Yao, Z.M. Electrochemical behaviour of chromium in acid solutions with H2S. Corros. Sci. 1999, 41, 773–788. [Google Scholar] [CrossRef]
- Smith, S.N. Current Understanding of Corrosion Mechanisms Due to H2S in Oil and Gas Productions Environments; Corrosion 2015, Paper No. 5485; NACE International: Houston, TX, USA, 2015. [Google Scholar]
- Depover, T.; Verbeken, K. The effect of TiC on the hydrogen induced ductility loss and trapping behavior of Fe-C-Ti alloys. Corros. Sci. 2016, 112, 308–326. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).