3. Results and Discussion
Table 1 lists the weight gains of Fe-(9, 19, 28, 37)Cr alloys due to corrosion at 700 and 800 °C for 70 h, which were measured using a microbalance before and after corrosion. Fe-9Cr always displayed the worst corrosion resistance, gaining excessive weight. For example, Fe-9Cr oxidized fast even in the N
2 gas through the reaction with impurities such as 3 ppm H
2O and 2 ppm O
2 in the N
2 gas (99.999% pure). Fe-9Cr oxidized faster in the N
2/H
2O gas than in the N
2 gas because of water vapor [
7]. Water vapor dissociates into oxygen and hydrogen, oxidizes the metal, and forms voids within the oxide scale according to the equation [
1,
3],
Fe-9Cr corroded the most seriously in N
2/H
2O/H
2S gas, because H
2S was much more harmful than H
2O. H
2S dissociates into hydrogen and sulfur. Sulfur forms non-protective metal sulfides according to the following equation:
Hydrogen, which is released from H
2S and H
2O, dissolves and ingresses into the alloy and the scale interstitially, generates lattice point defects, forms hydrogen clusters and voids, causes hydrogen embrittlement, produces volatile hydrated species, and accelerates cracking, spallation and fracture of the scale. Hence, no metals are resistant to H
2O/H
2S corrosion. As listed in
Table 1, Fe-(19, 29, 37)Cr displayed much better corrosion resistance in N
2 and N
2/3.2%H
2O with weight gains of 1–2 mg/cm
2 than Fe-9Cr. Fe-(19, 29, 37)Cr formed 0.3- to 1.3-μm-thick, adherent oxide scales. However, even Fe-(19, 29, 37)Cr failed in N
2/3.1%H
2O/2.42%H
2S with large weight gains, forming non-adherent, fragile sulfide scales as thick as 35–750 μm. This scale failure made the weight gains measured in N
2/H
2O/H
2S gas inaccurate. In N
2/H
2O/H
2S gas, the amount of local cracking, spallation and void formation in the scale varied for each test run. Although the accurate measurement of weight gains in N
2/H
2O/H
2S gas was impossible, it was clear that weight gains due to scaling decreased sharply with the addition of Cr.
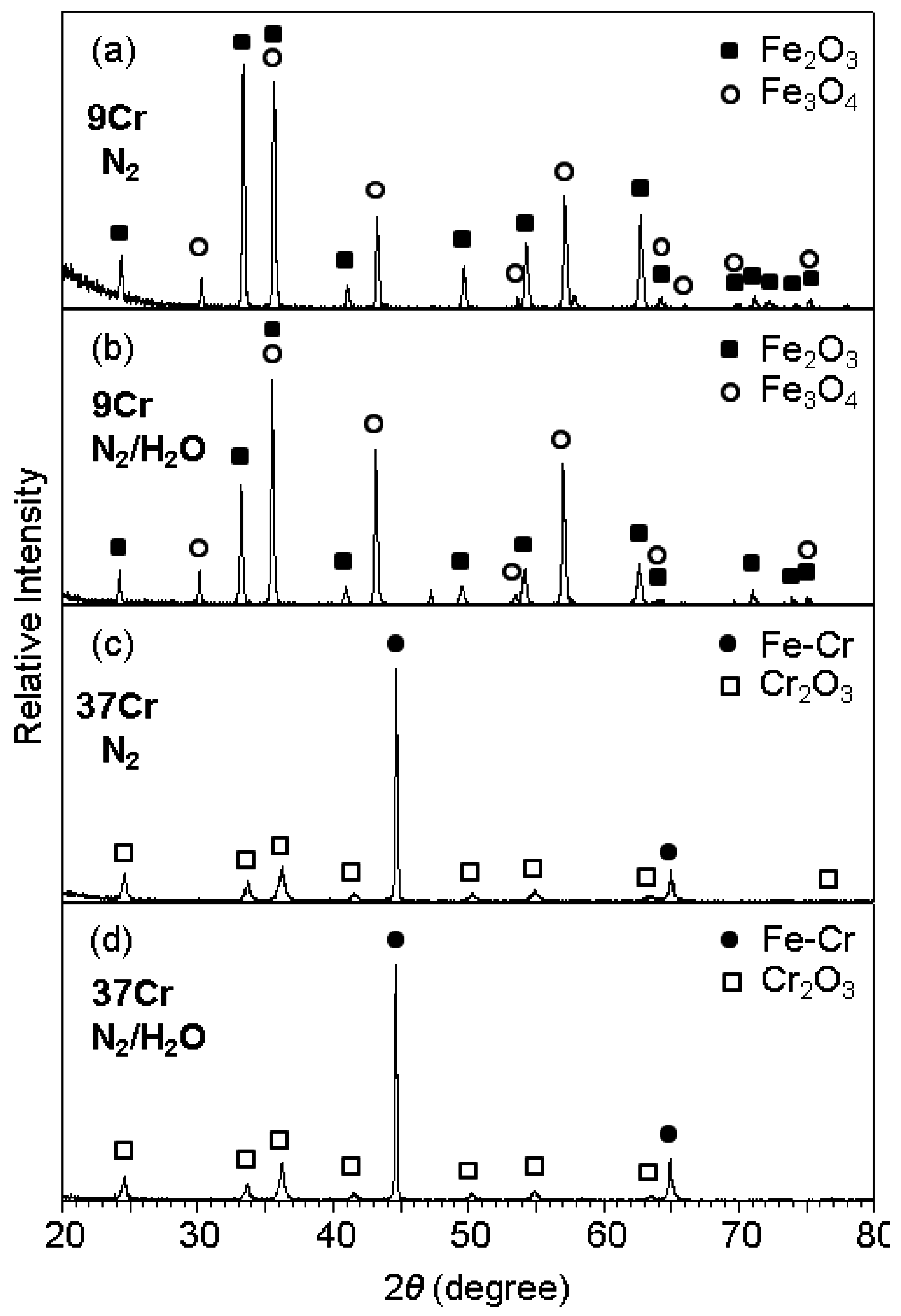
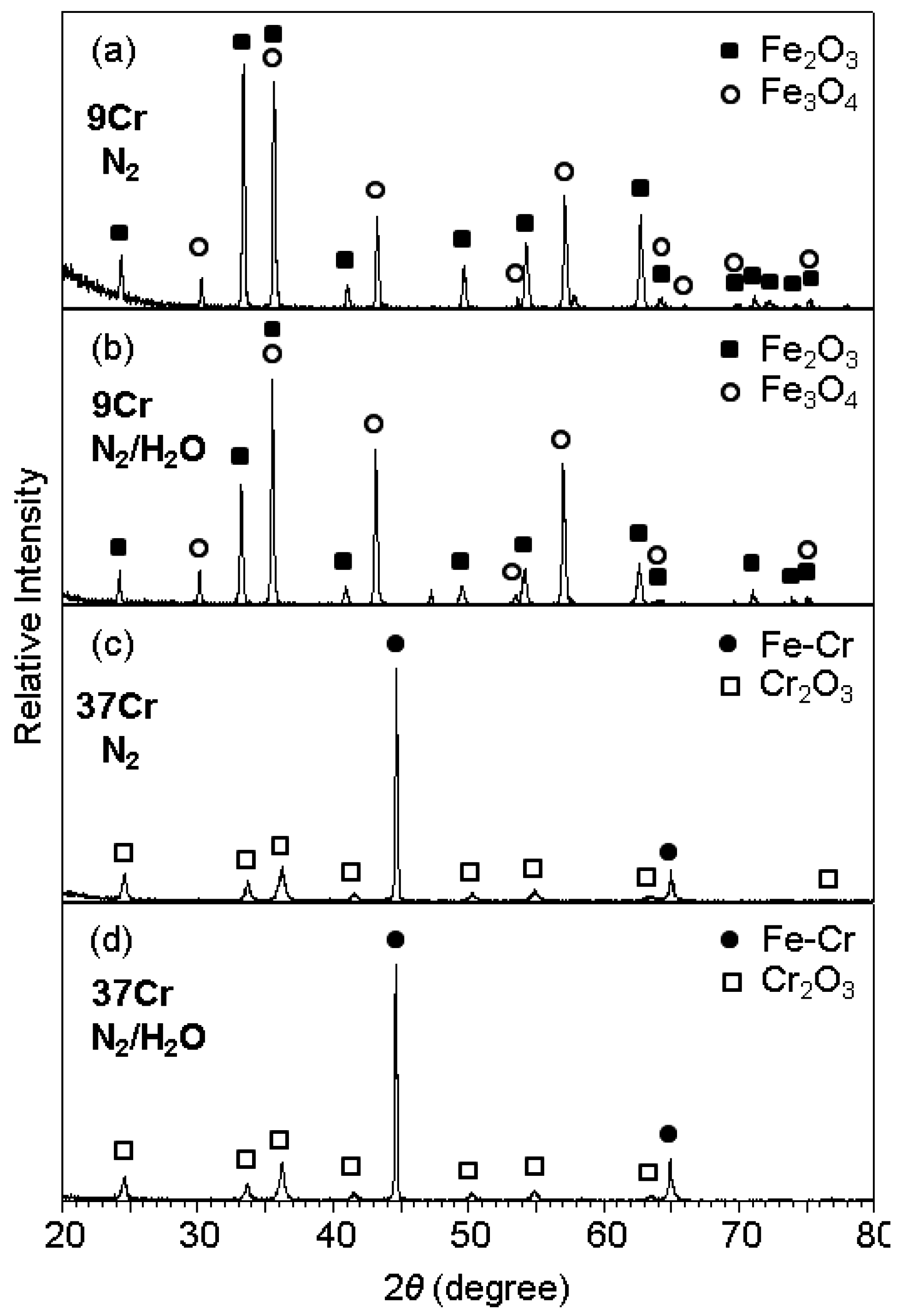
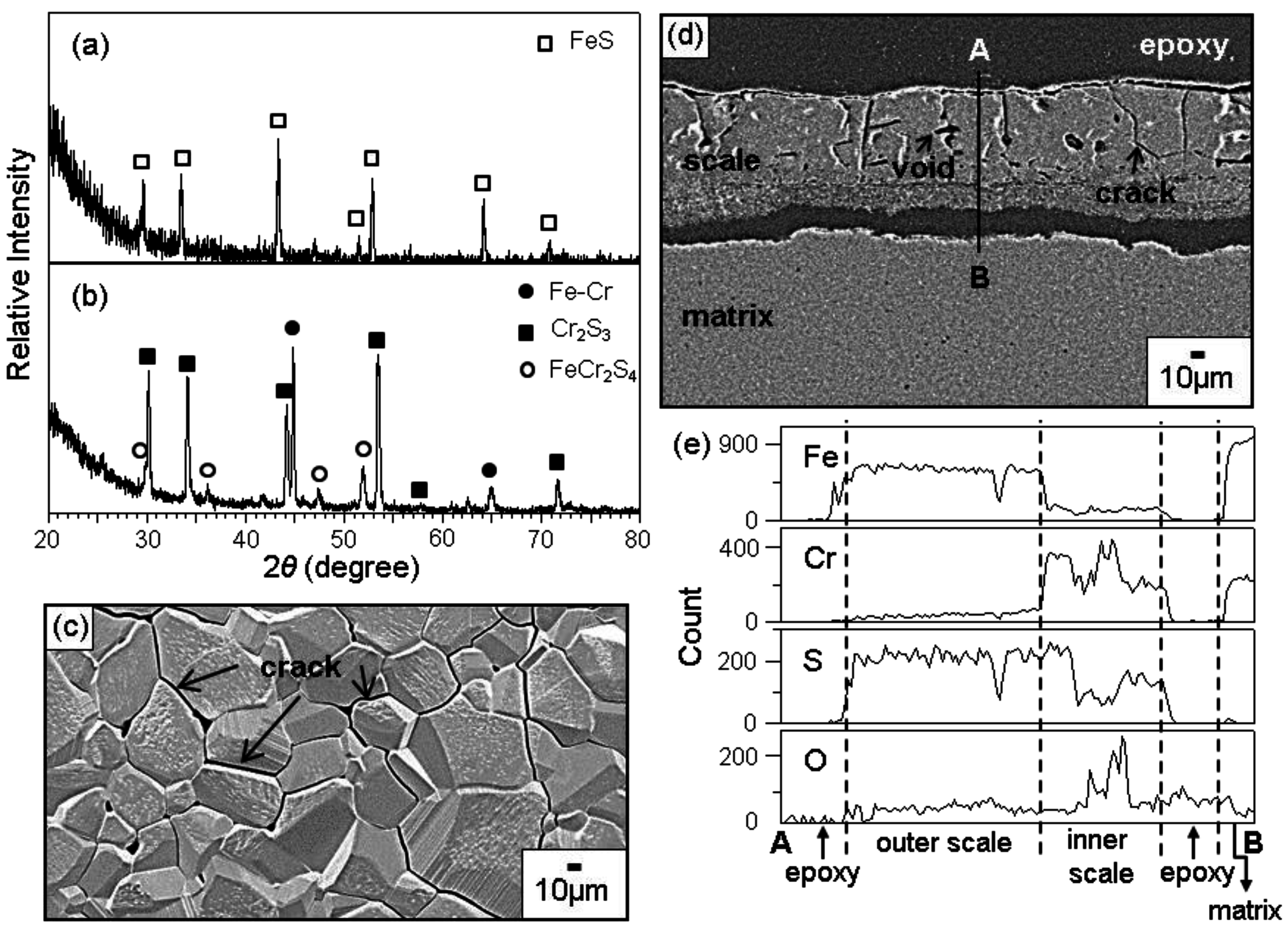
Figure 2 shows the XRD patterns of scales formed after corrosion at 800 °C for 70 h. The corrosion of Fe-9Cr in N
2 and N
2/H
2O resulted in the formation of Fe
2O
3 and Fe
3O
4, as shown in
Figure 2a,b. Oxide scales formed on Fe-9Cr in N
2 and N
2/H
2O were 90 and 100 μm thick, respectively. Since X-rays could not penetrate such thick oxide scales, FeO and Cr-oxides such as FeCr
2O
4, which might form next to the alloy [
1], were absent in
Figure 2a,b. In contrast, Fe-(19, 28, 37)Cr alloys oxidized at much slower rates in N
2 and N
2/H
2O than Fe-9Cr alloy, as listed in
Table 1. Fe-(19, 28, 37)Cr alloys formed the protective Cr
2O
3 scale, as typically shown in
Figure 2c,d. Here, the Fe-Cr peaks were strong owing to the thinness of the oxide scales. In Fe-(19, 28, 37)Cr alloys, Cr was dissolved in the α-Fe matrix.
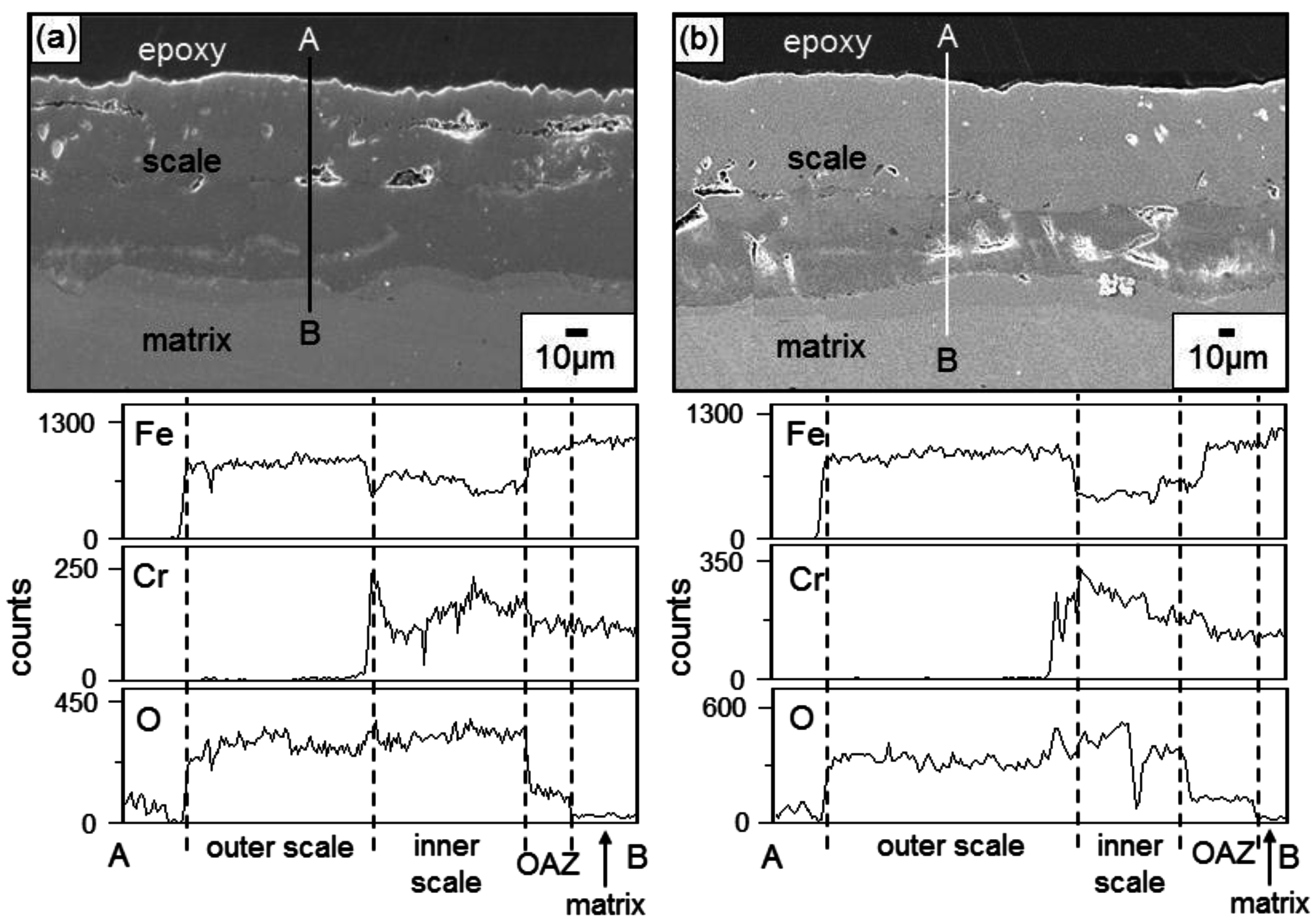
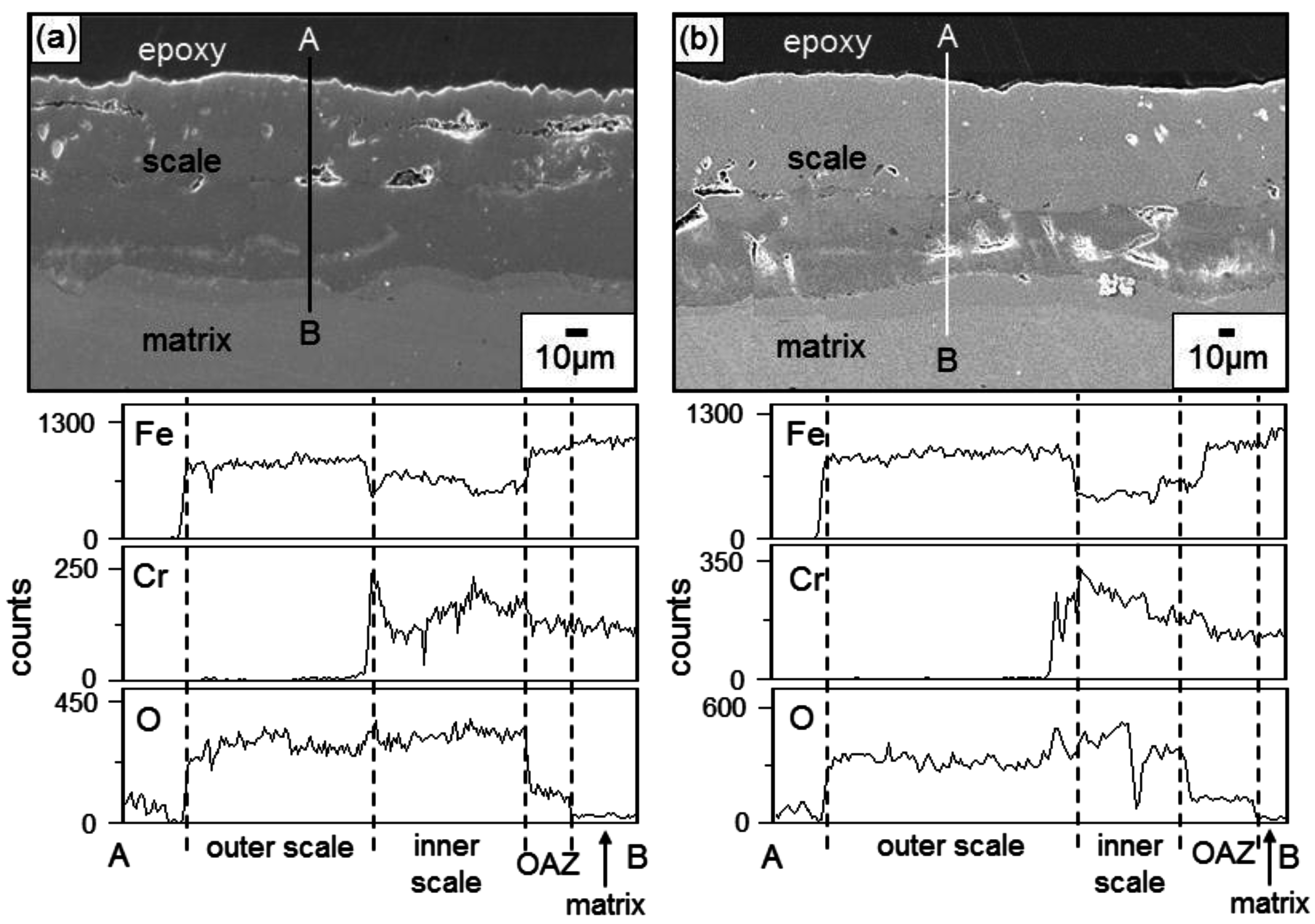
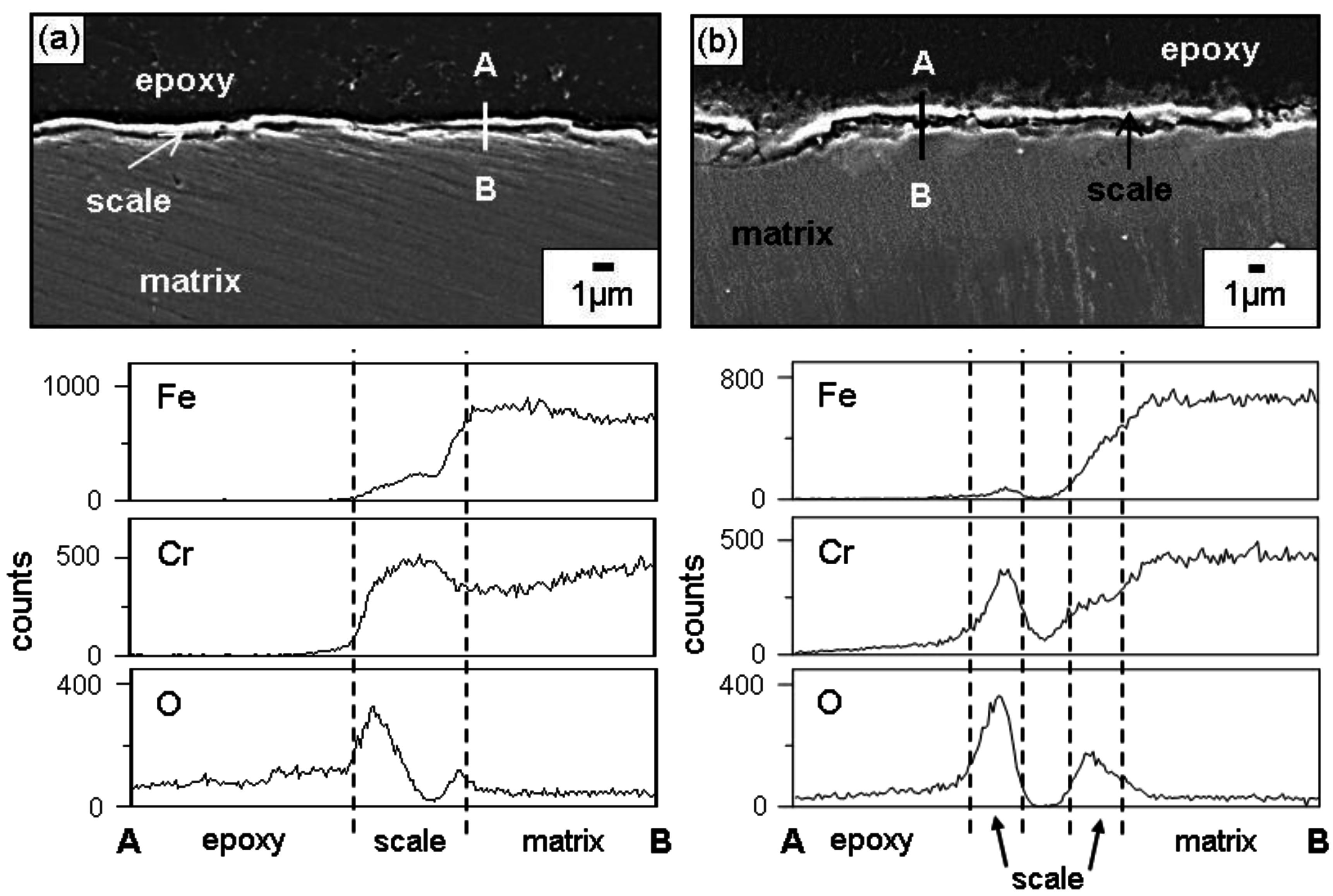
Figure 3 shows the EPMA analytical results on the scales formed on Fe-9Cr after corrosion at 700 °C for 70 h. The oxide scales that formed after corrosion in N
2 and N
2/H
2O were about 90 and 140 μm thick, respectively. The scale morphology and elemental distribution in N
2 gas were similar to those in N
2/H
2O gas, as shown in
Figure 3, indicating that the same oxidation mechanism operated in N
2 and N
2/H
2O gas. Voids were sporadically scattered in both oxide scales, below which the oxygen-affected zone (OAZ) existed. Voids formed owing to the volume expansion during scaling, hydrogen released from the water vapor, and the Kirkendall effect arose due to the outward diffusion of cations during scaling. In both oxide scales, the outer layer consisted of iron oxides, while the inner layer consisted of (Fe,Cr) mixed oxides. This indicated that Fe
2+ and Fe
3+ ions were more mobile than Cr
3+ ions. The oxidation in N
2 and N
2/H
2O gas was mainly controlled by the outward diffusion of iron ions through the inner (Fe,Cr) mixed oxide layer. Iron oxidized preferentially in N
2 and N
2/H
2O gas because iron is the base element and its oxide, FeO, is a non-stoichiometric compound with a relatively fast growth rate.
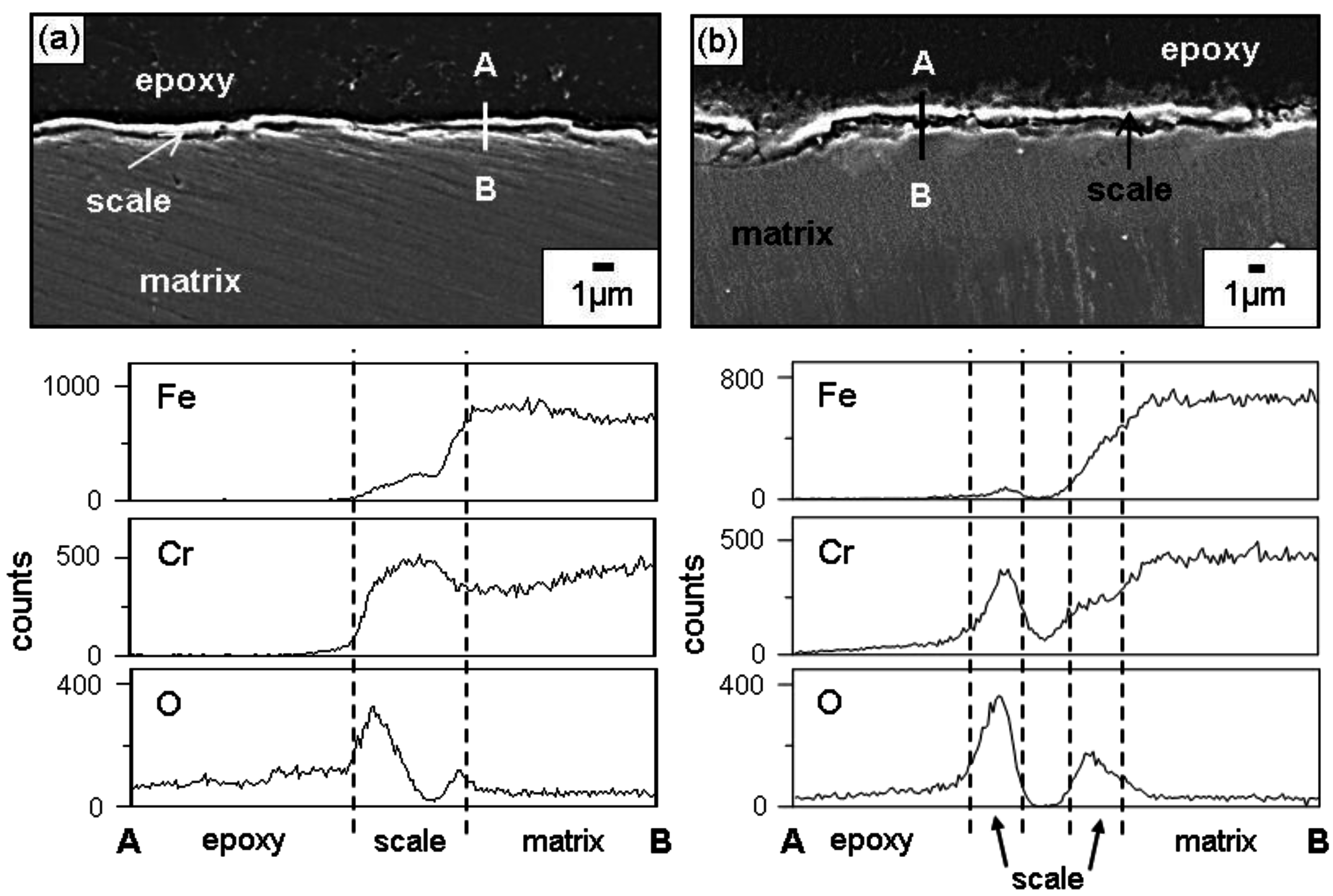
Figure 4 shows the EPMA analytical results on the scales formed on Fe-37Cr after corrosion at 700 °C for 70 h. The oxide scales that formed after corrosion in N
2 and N
2/H
2O were about 0.6 and 1.1 μm thick, respectively. In N
2 and N
2/H
2O gas, the Cr
2O
3 scale formed (
Figure 2c,d), in which Fe was dissolved (
Figure 4). The complete dissolution of Fe
2O
3 in Cr
2O
3 is possible, because Cr
2O
3 and Fe
2O
3 have the same rhombohedral structure [
8]. Like Fe-37Cr, Fe-(19, 28)Cr also formed a thin Cr
2O
3 scale containing some Fe when they corroded in N
2 and N
2/H
2O gas. Once the thin but protective Cr
2O
3 scale formed, the outward diffusion of iron ions was suppressed so that good corrosion resistance was achieved.
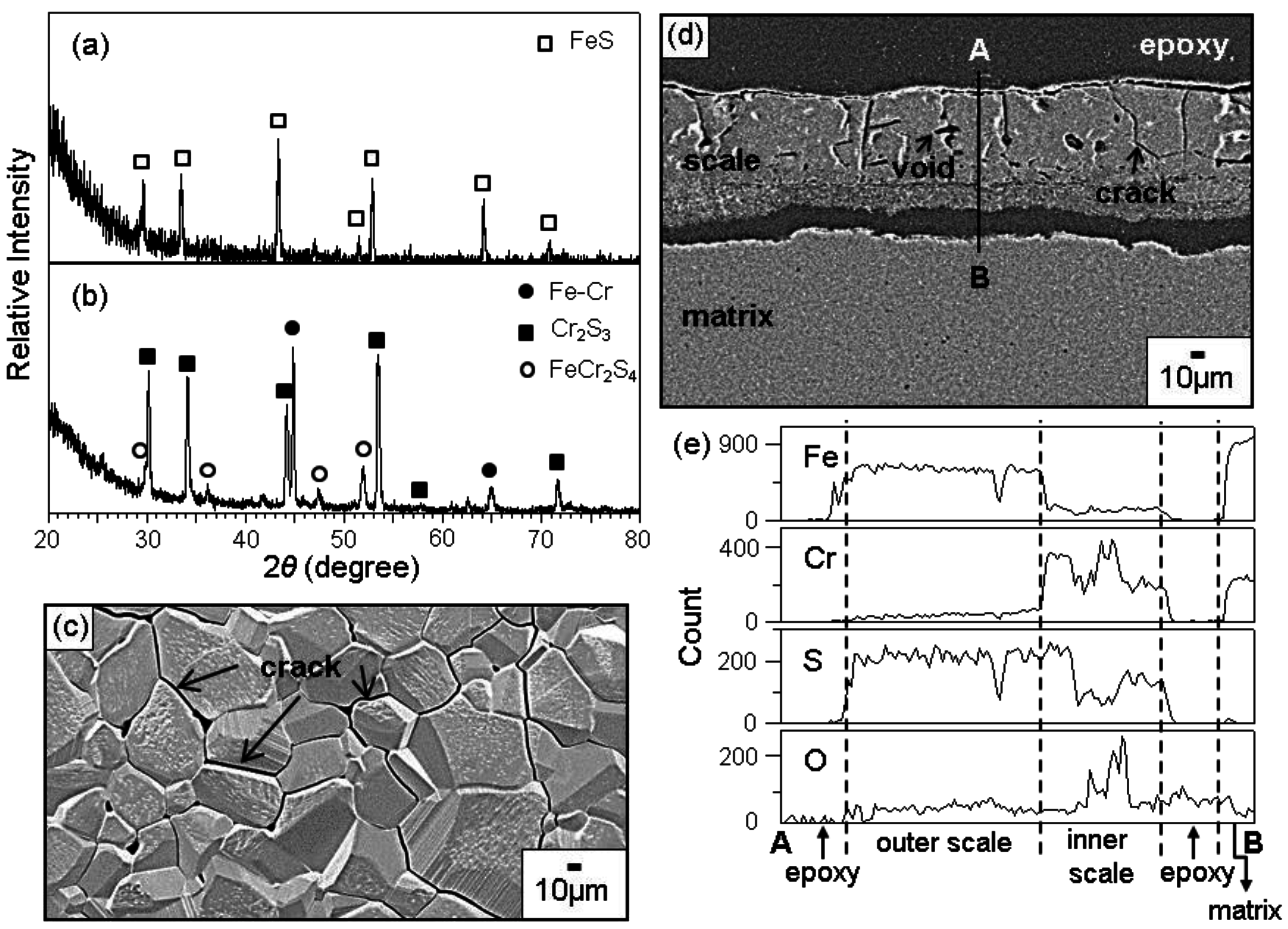
In N
2/3.1%H
2O/2.42%H
2S gas, Fe-(9-37)Cr alloys could not form Cr
2O
3, and corroded fast, as typically shown in
Figure 5. The scales formed on Fe-(9, 19, 28, 37)Cr alloys consisted primarily of the outer FeS layer (
Figure 5a), and the inner Cr
2S
3 layer containing some FeCr
2S
4 (
Figure 5b). Since FeS grows fast owing to its high non-stoichiometry, outer FeS grains were coarser than the inner (Cr
2S
3, FeCr
2S
4) mixed grains. In
Figure 5c, cracks propagated inter- and trans-granularly due mainly to the excessive growth stress generated in the thick outer scale. The scale shown in
Figure 5d was about 100 μm thick, and had cracks and voids. A small amount of Cr was dissolved in the outer FeS layer (
Figure 5e). The preferential sulfidation of iron in the outer FeS layer decreased the sulfur potential underneath, and thereby increased the oxygen potential in the inner Cr
2S
3-rich layer, leading to the incorporation of oxygen in the inner Cr
2S
3-rich layer. FeS is a p-type metal-deficit compound, which grows fast by the outward diffusion of Fe
2+ ions [
5,
9,
10]. Its defect chemical equation is as follows.
here, O
O, h
· and
mean the O atom on the O site, the electron hole in the valence band with a + 1 charge, and the iron vacancy with a − 2 charge. The defect chemical reaction for the dissolution of Cr
2S
3 in FeS is as follows.
Hence, the doping of Cr
3+ ions would increase the concentration of iron vacancies, leading to the enhancement of the FeS growth. Oxygen was incorporated in the inner Cr
2S
3-rich layer (
Figure 5e). However, no oxides were detected in
Figure 5b, because their amount was small or oxygen was dissolved in the sulfide scales. Grains in the inner layer were fine owing to the nucleation and growth of Cr
2S
3, together with some FeCr
2S
4 and probably some oxides. In N
2/3.1%H
2O/2.42%H
2S gas, Fe-(9, 19, 28, 37)Cr alloys sulfidized preferentially owing to the high sulfur potential in the test gas.



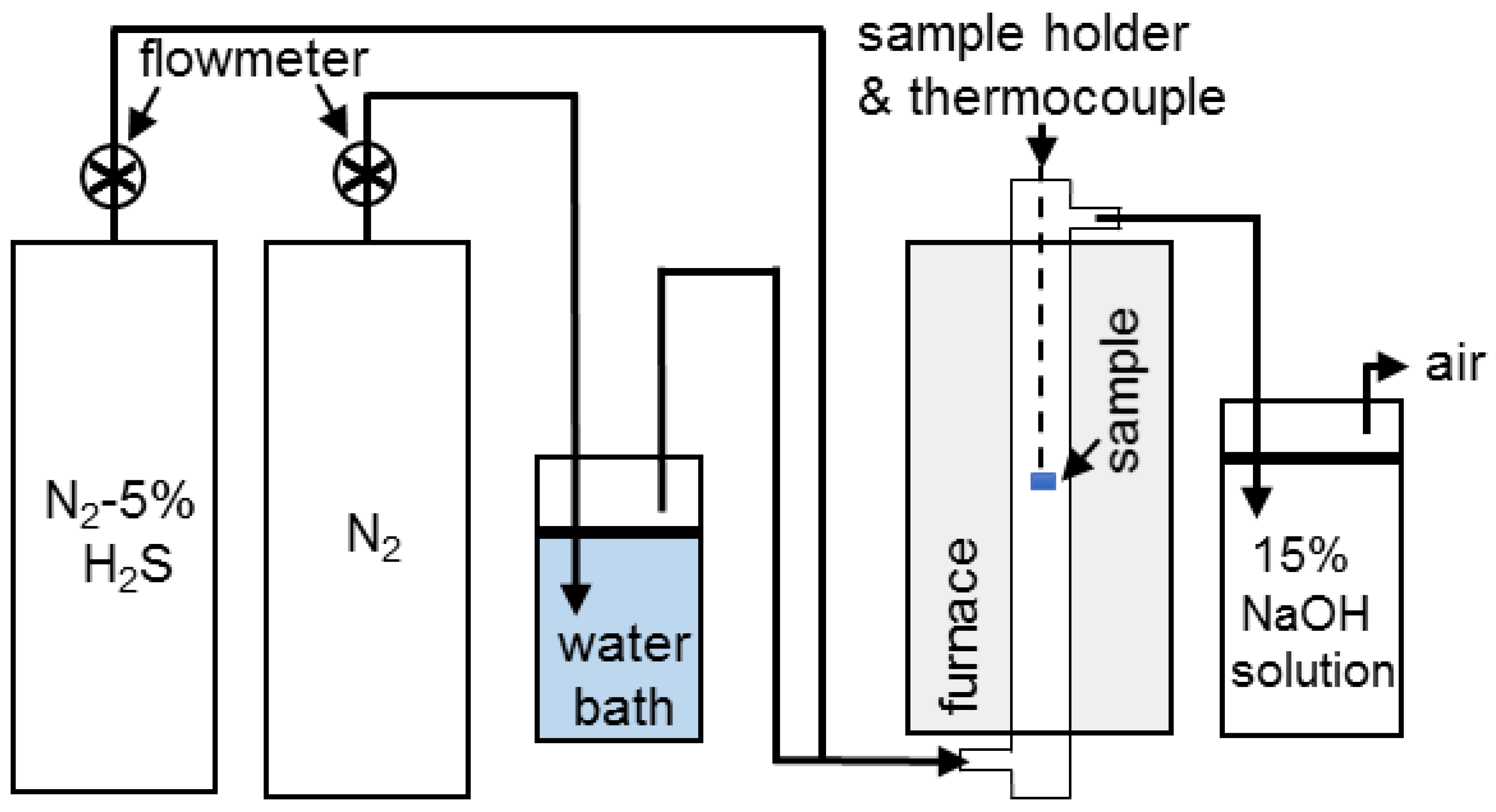

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}