
Separation of Rhenium from Lead-Rich Molybdenite Concentrate via Hydrochloric Acid Leaching Followed by Oxidative Roasting



Abstract
:1. Introduction
2. Experimental Section
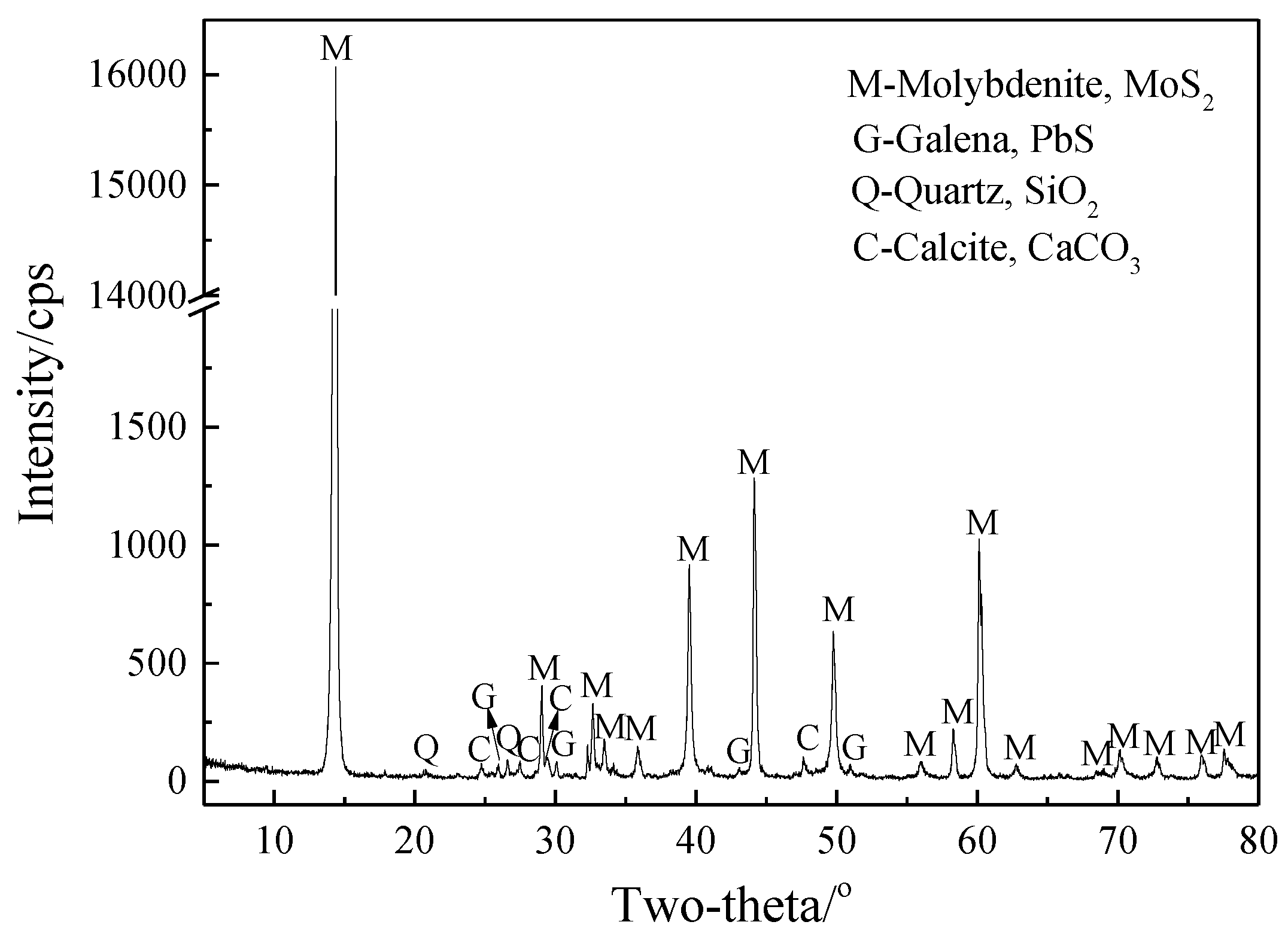
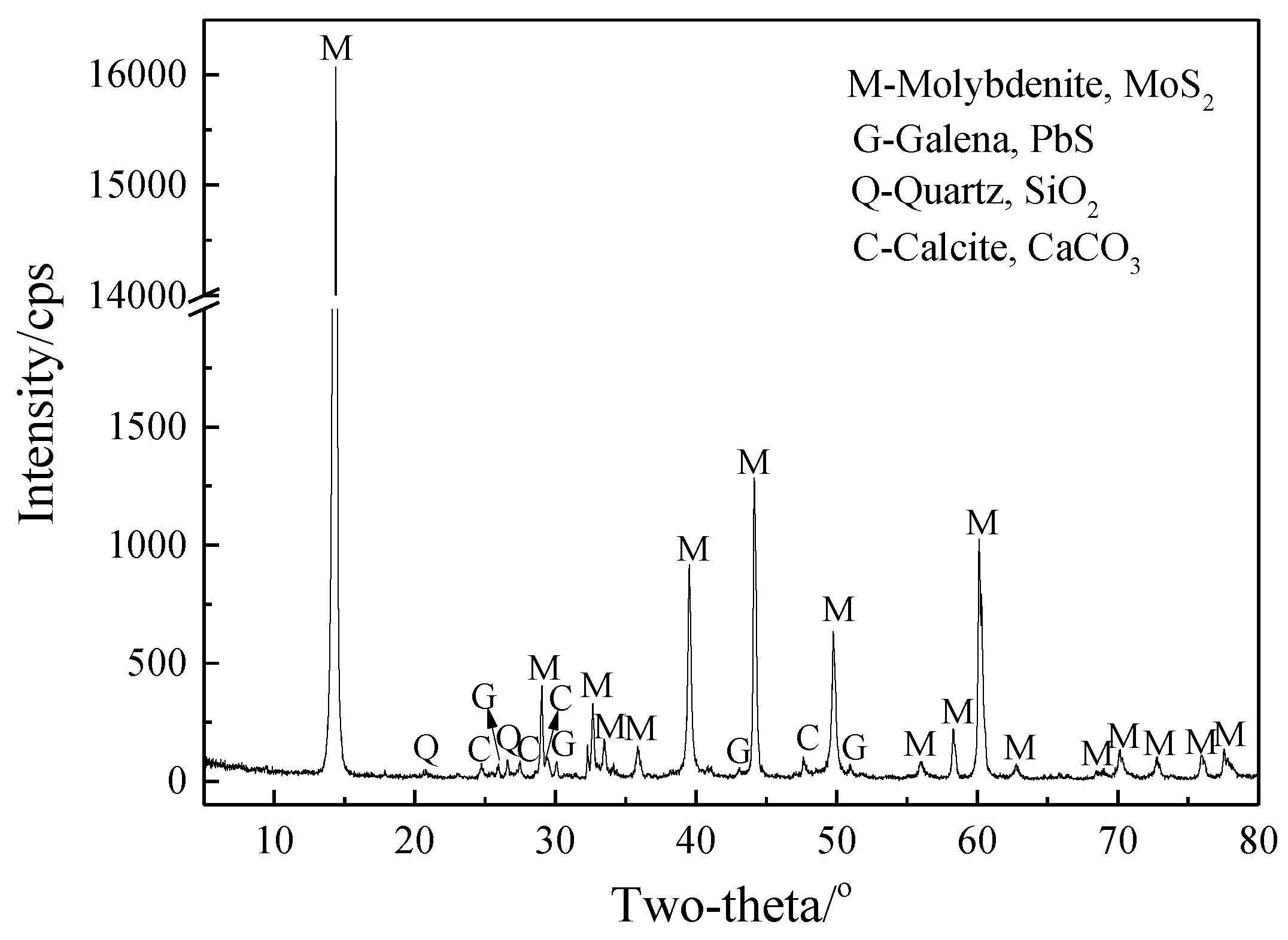
2.1. Materials
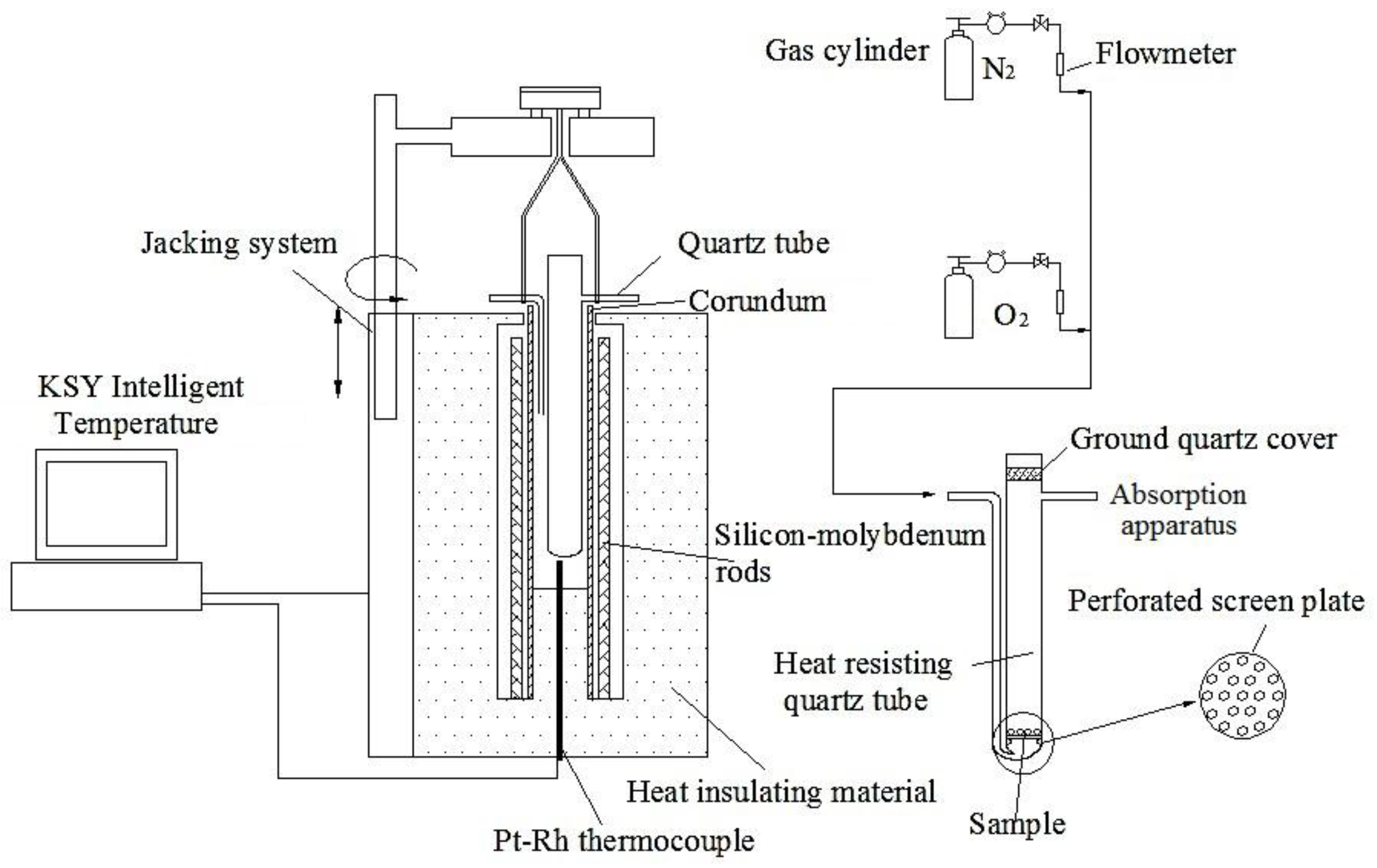
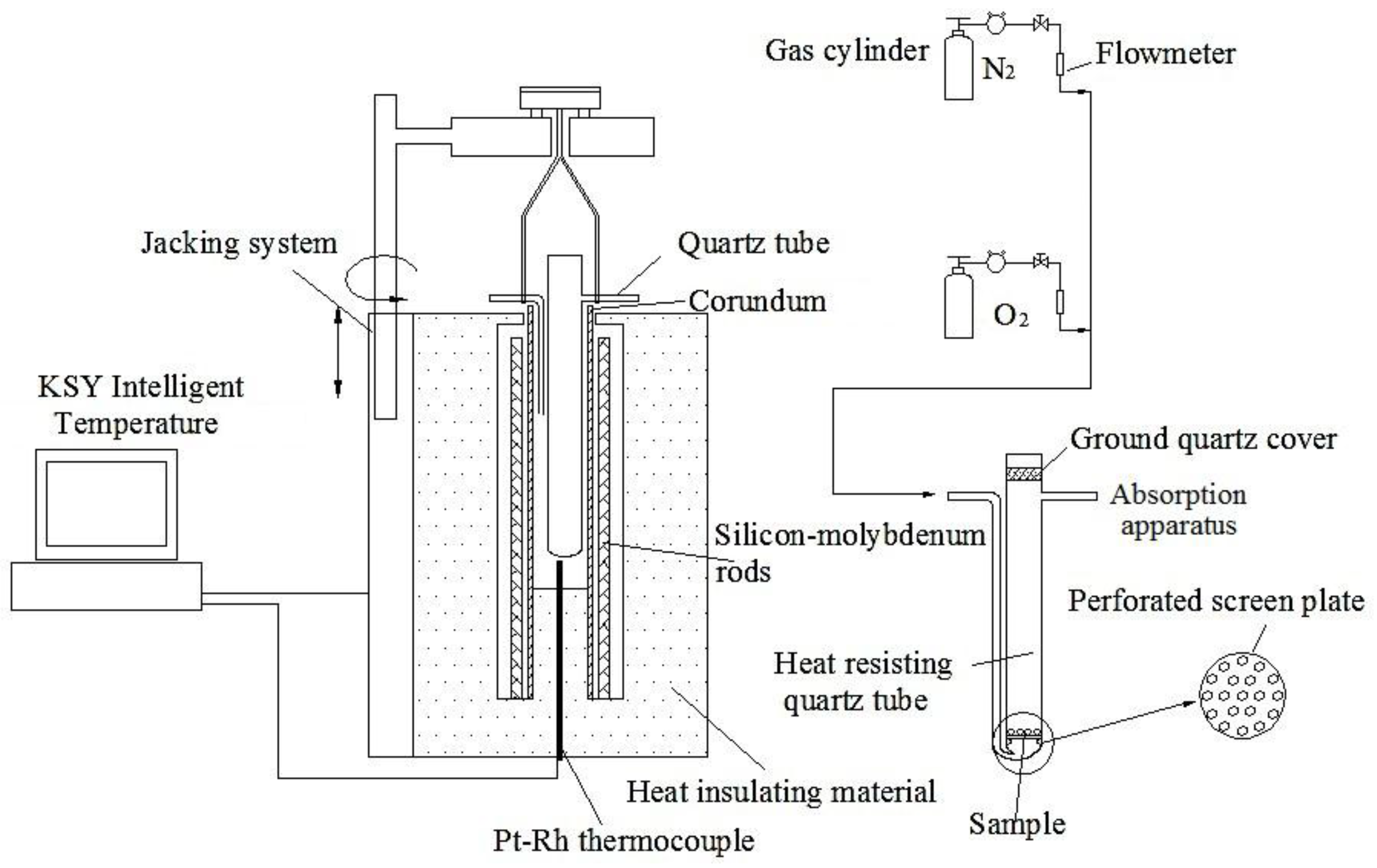
2.2. Experimental Procedure
2.3. Analytical Method
3. Results and Discussion
3.1. Removal of Pb by Acid Leaching with HCl
3.1.1. Chemistry of HCl Leaching
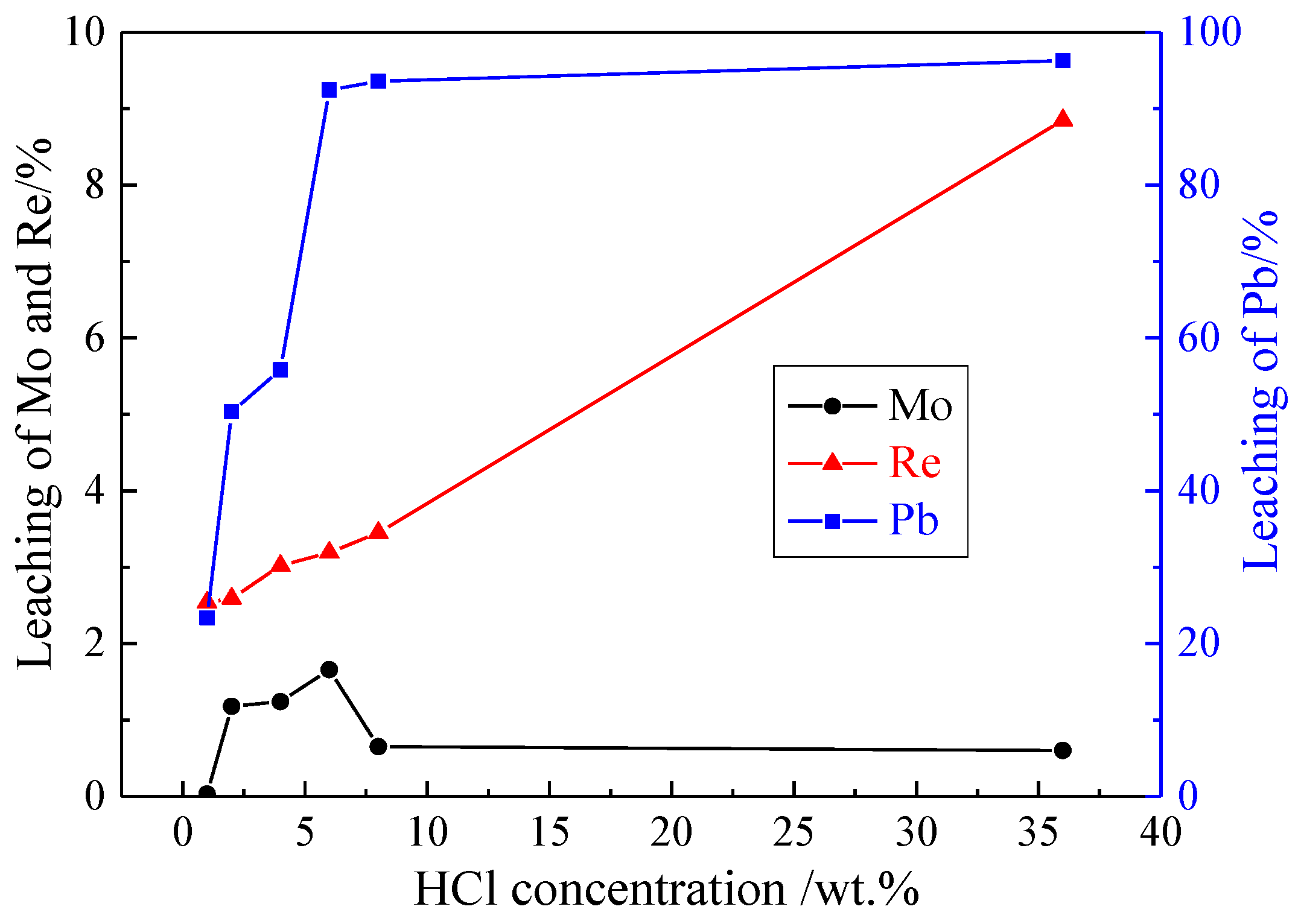
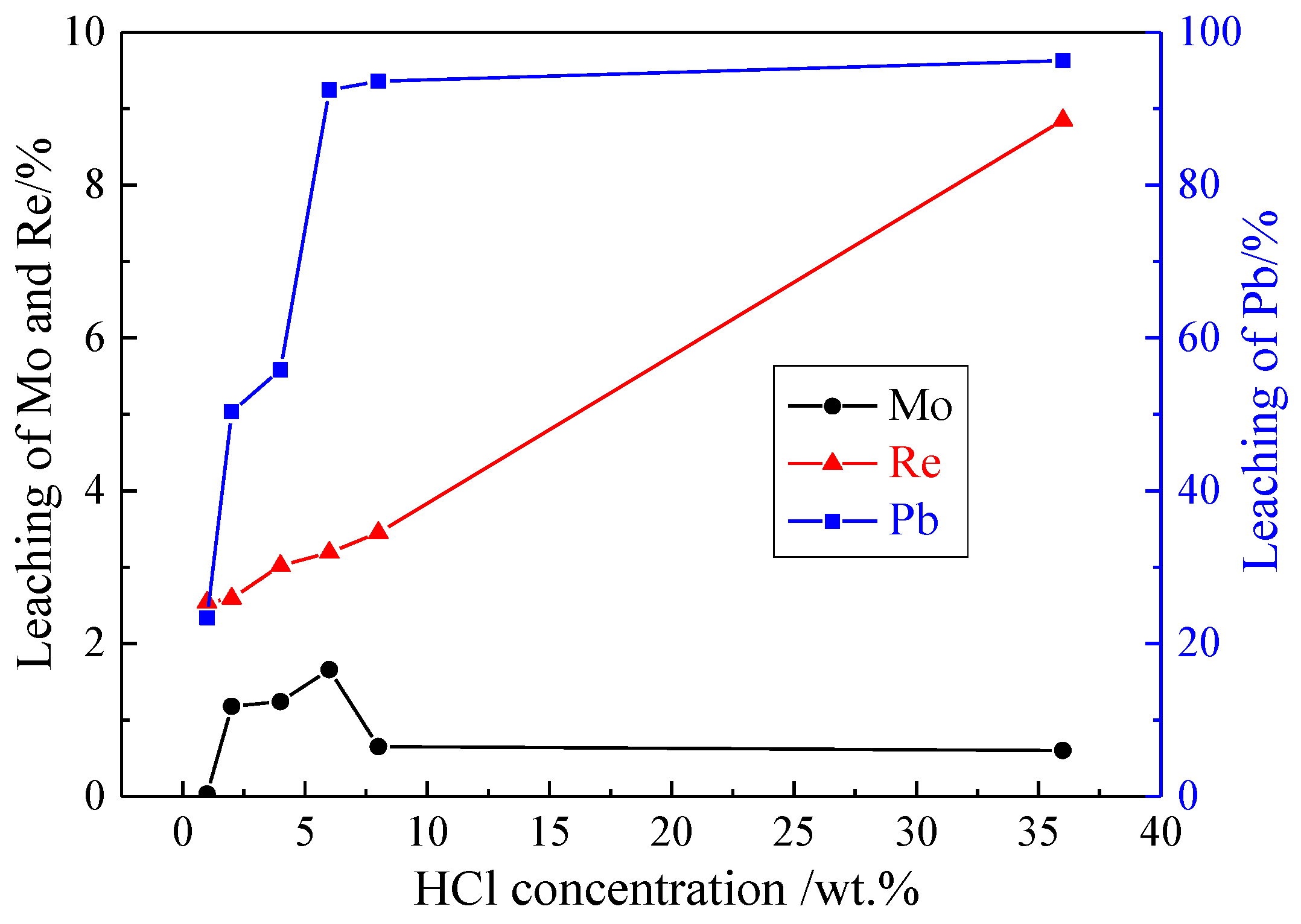
3.1.2. Effect of HCl Concentration
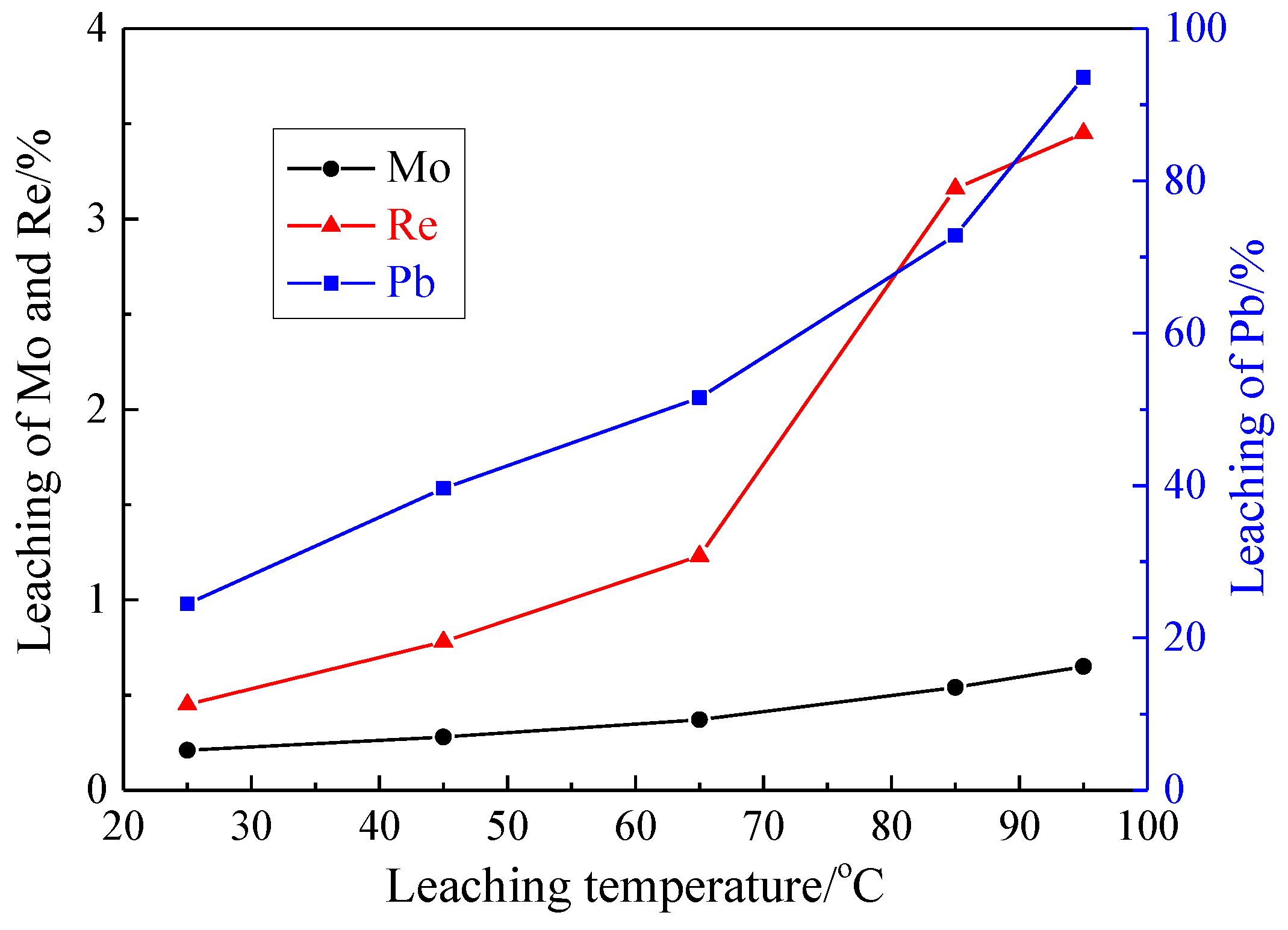
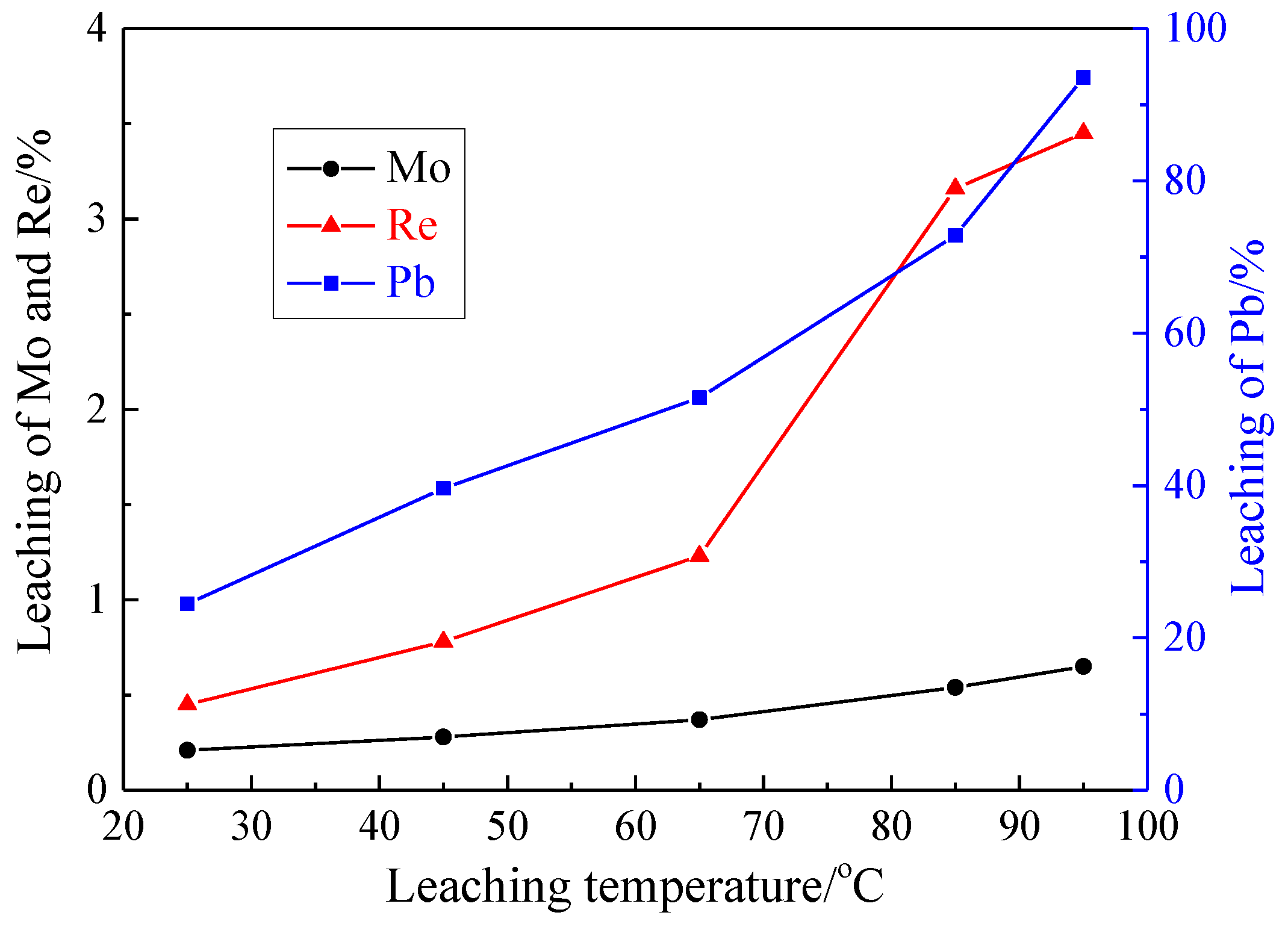
3.1.3. Effect of Leaching Temperature
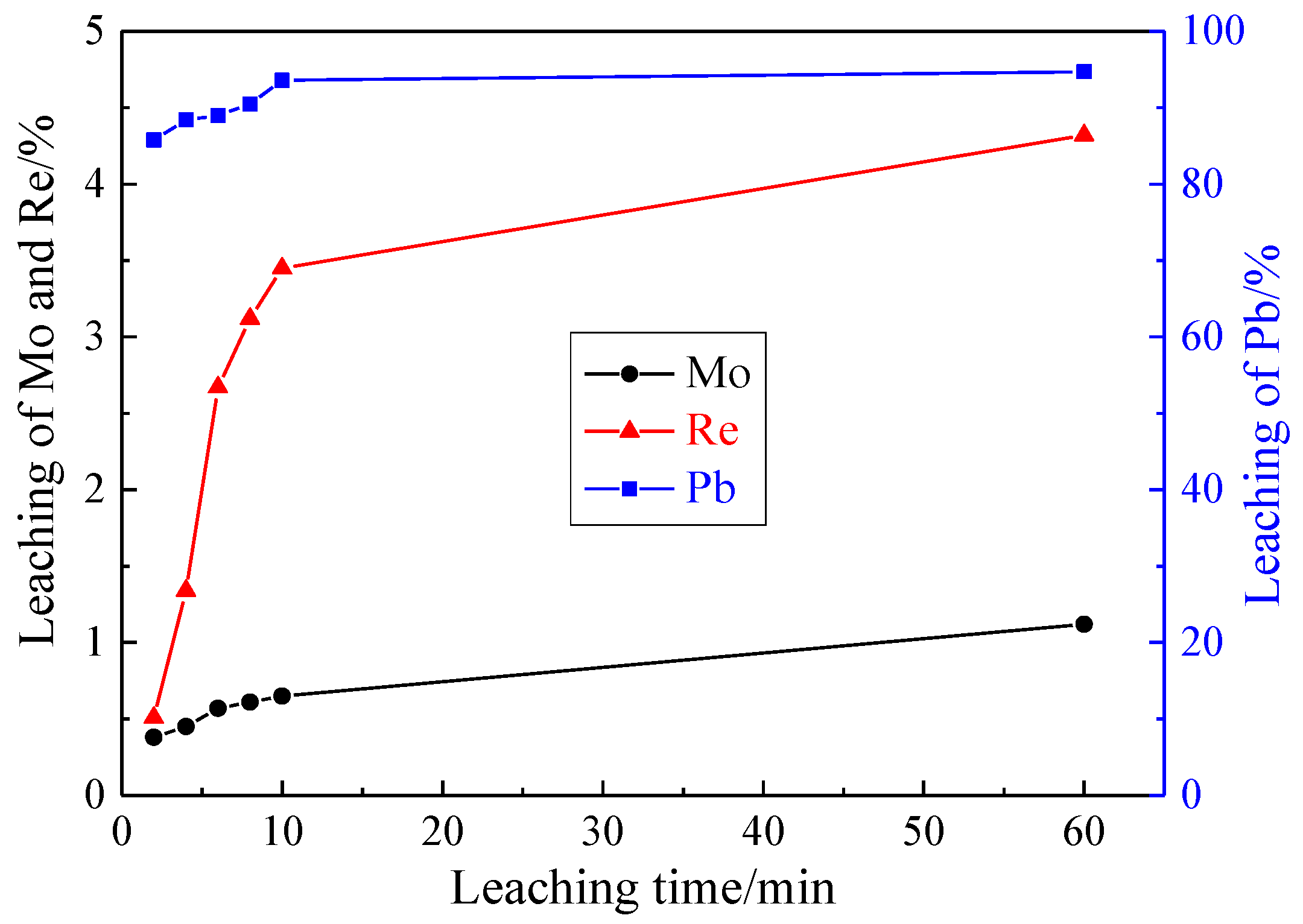
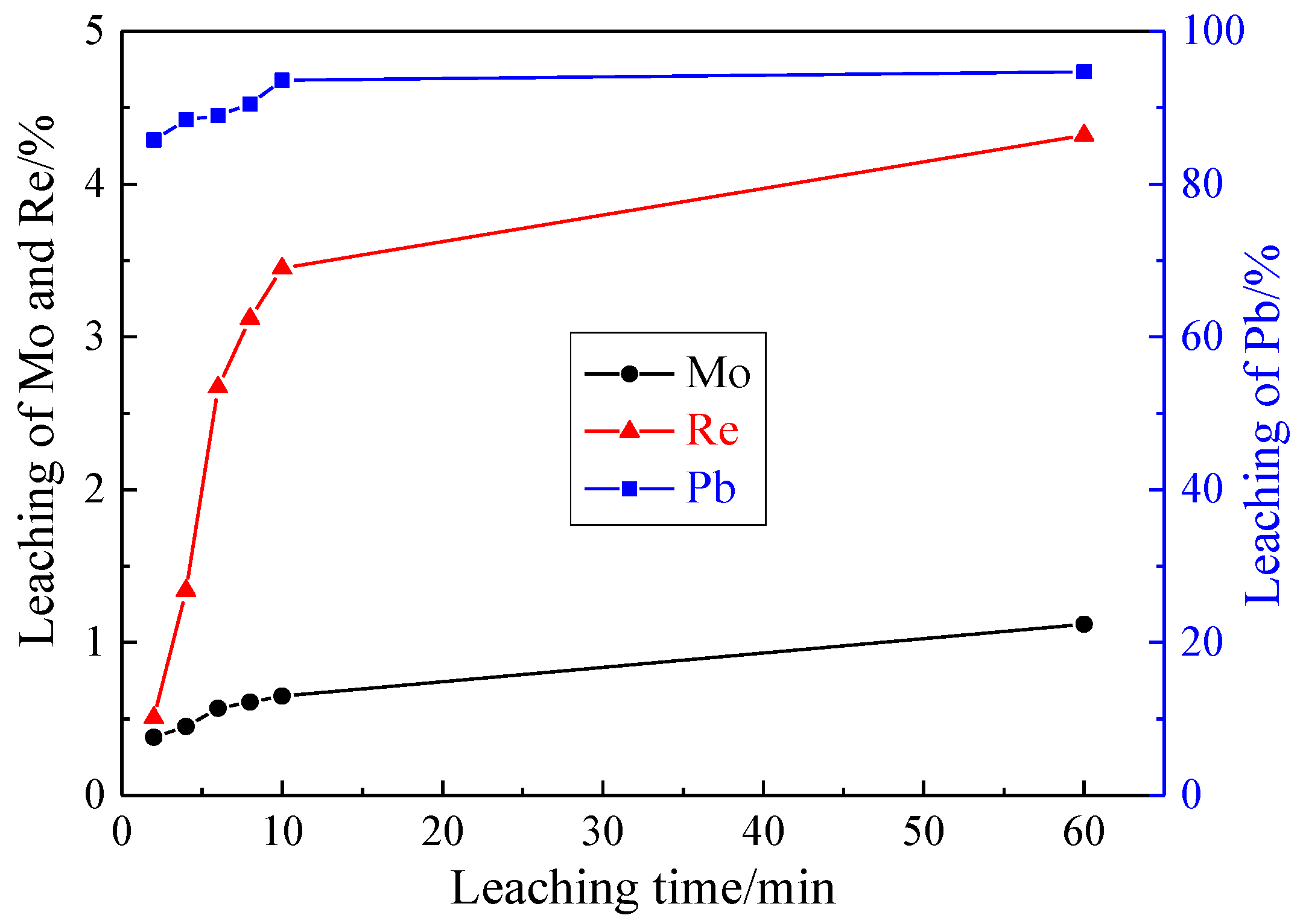
3.1.4. Effect of Leaching Time
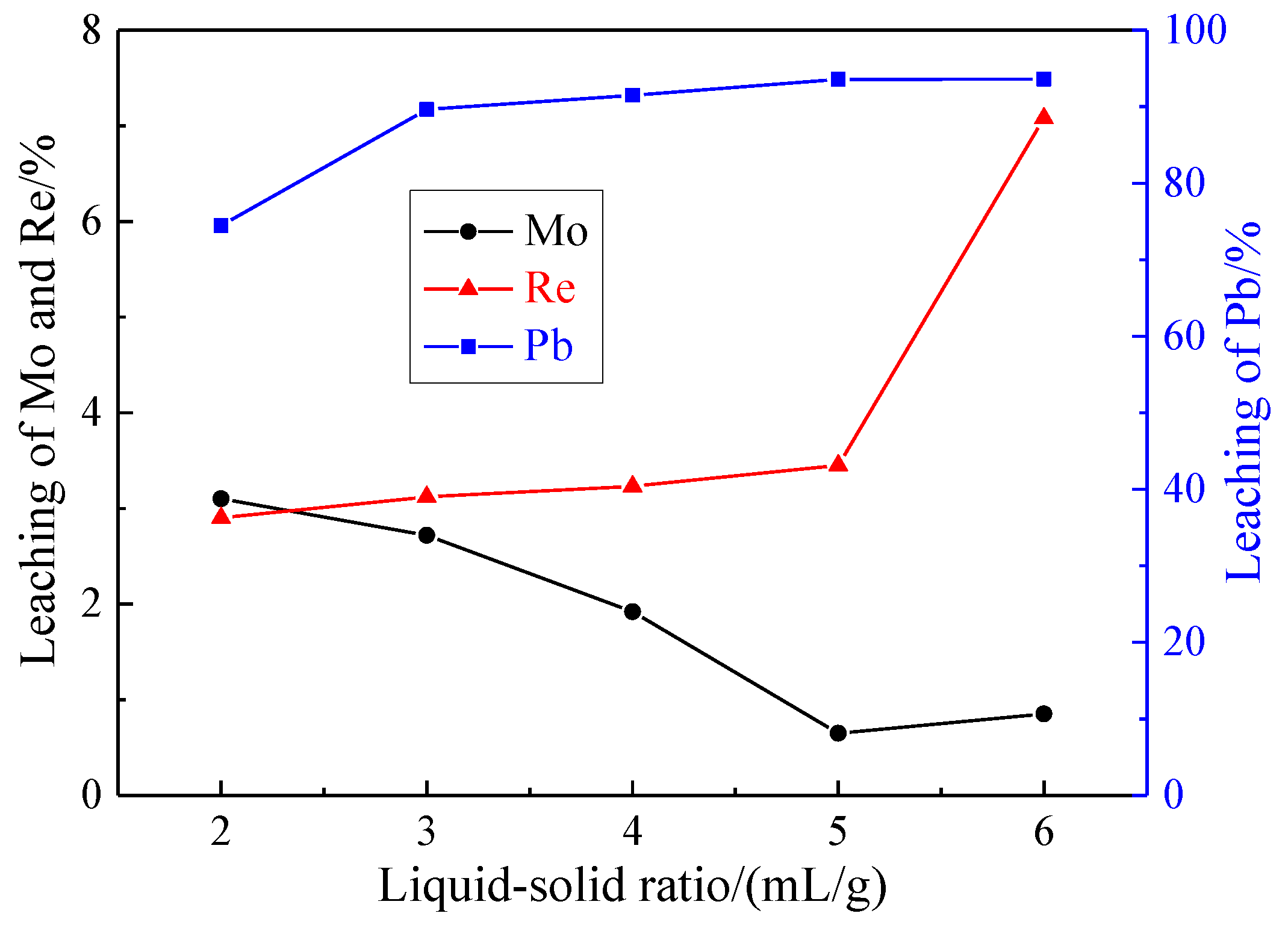
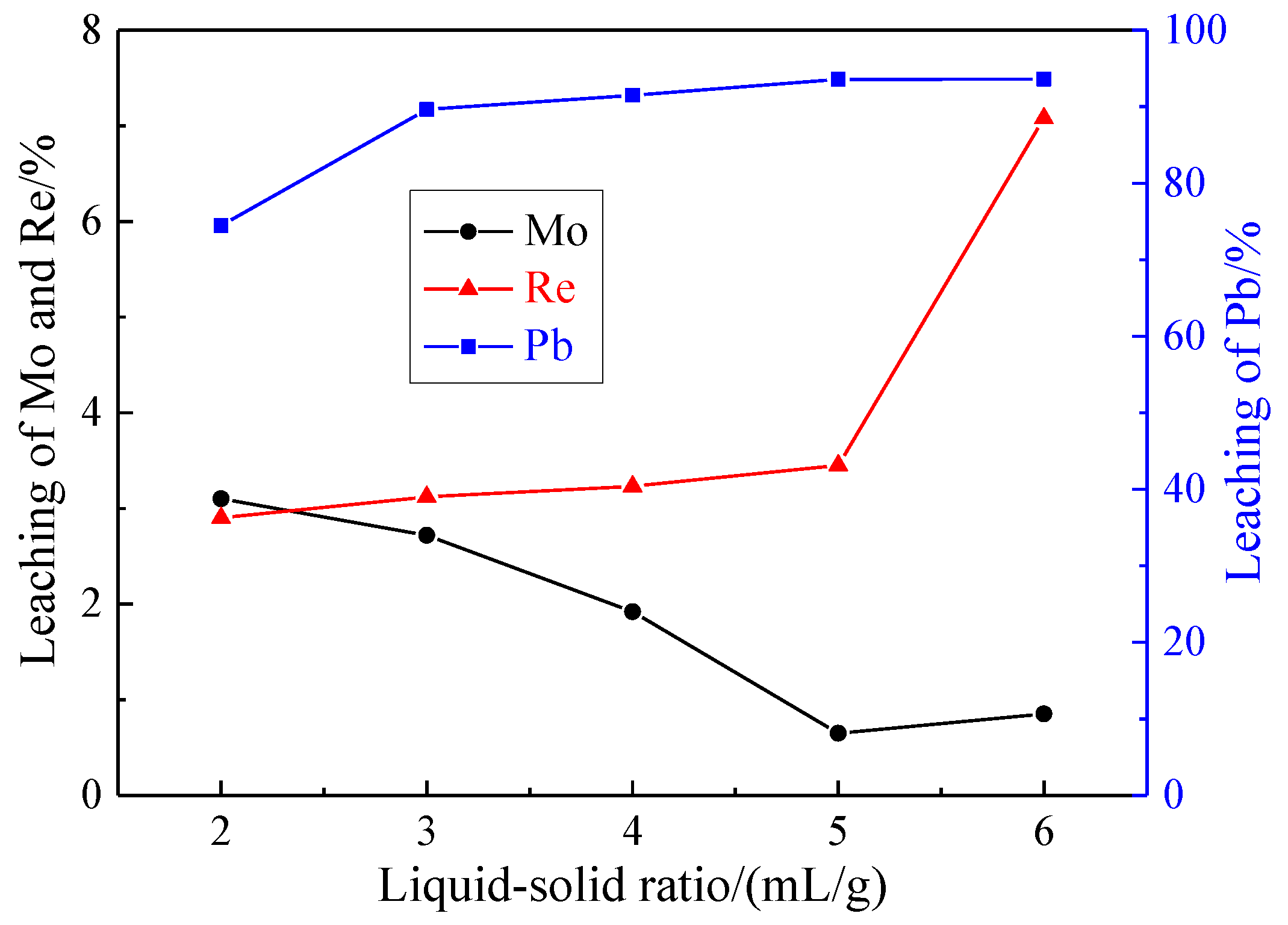
3.1.5. Effect of Liquid-Solid Ratio
3.2. Separation of Rhenium by Oxidative Roasting
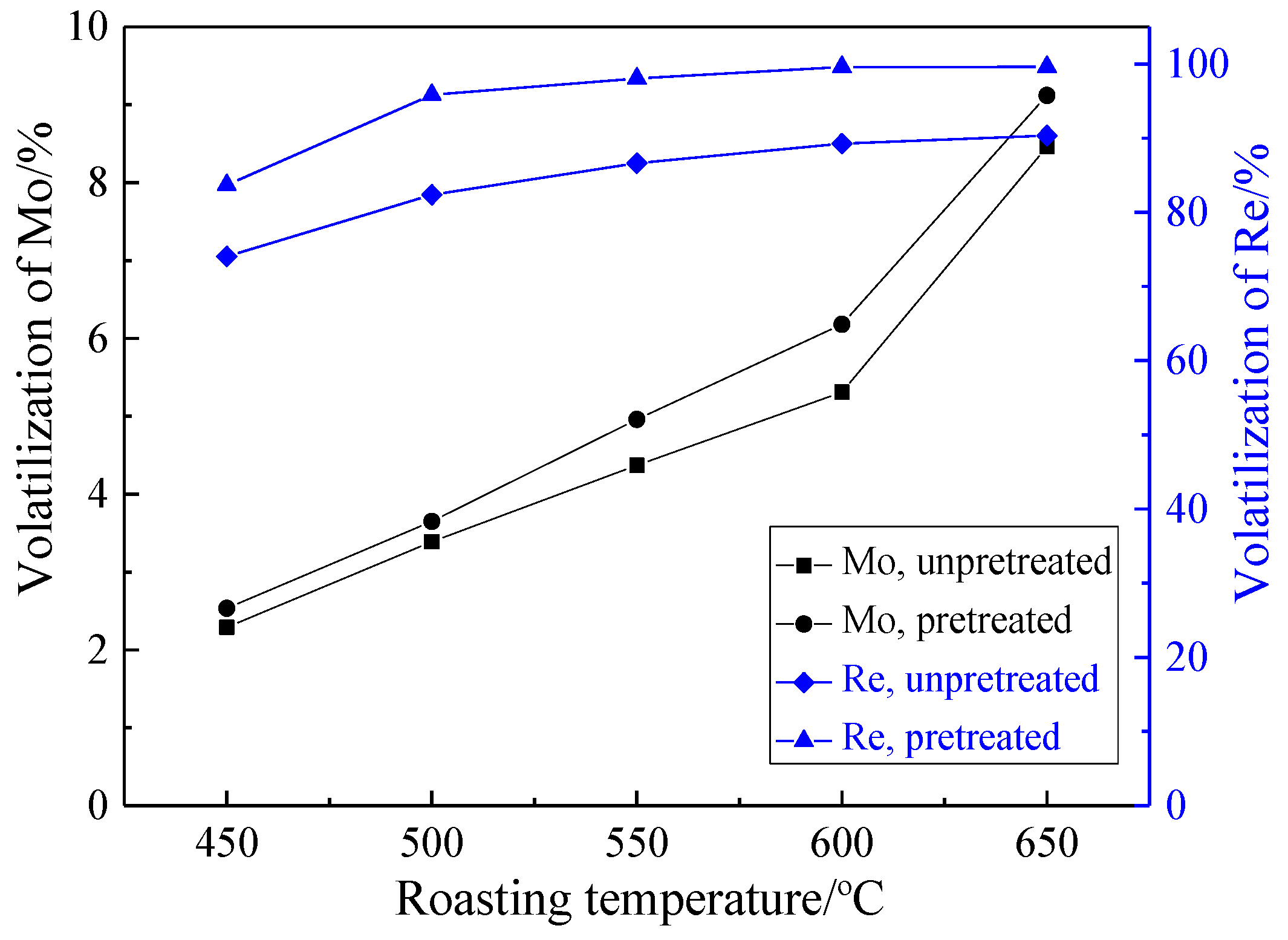
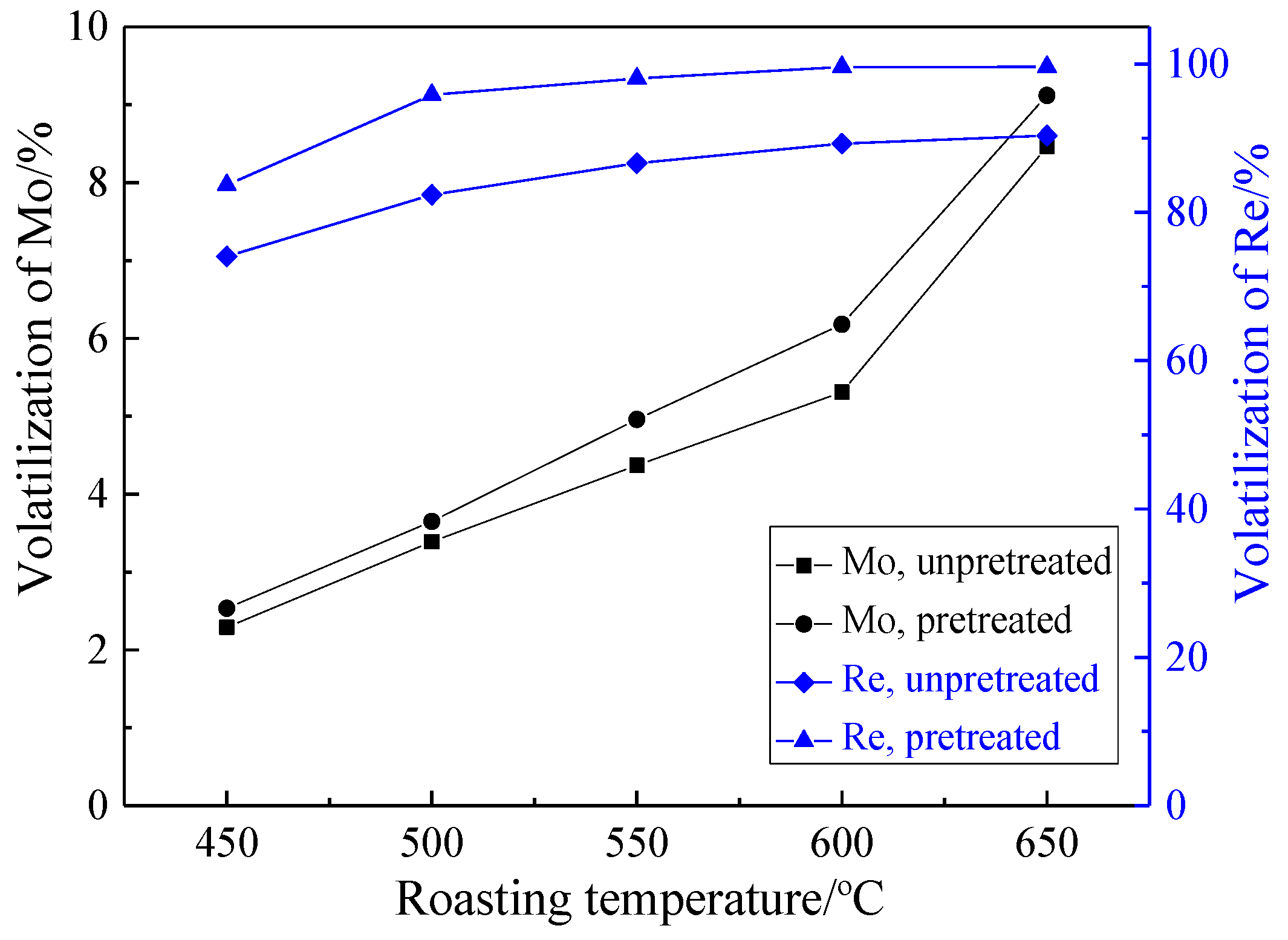
3.2.1. Effect of Roasting Temperature
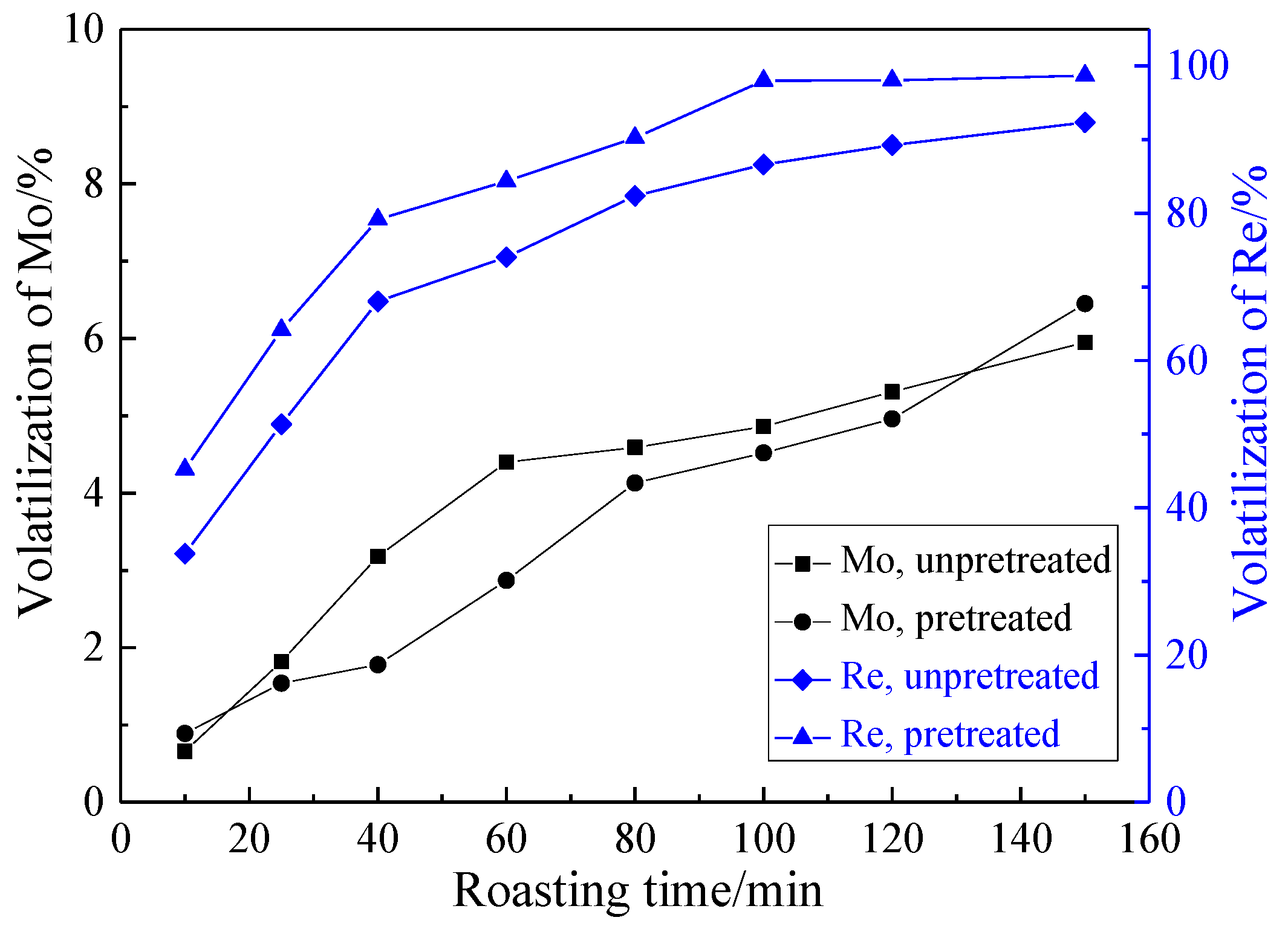
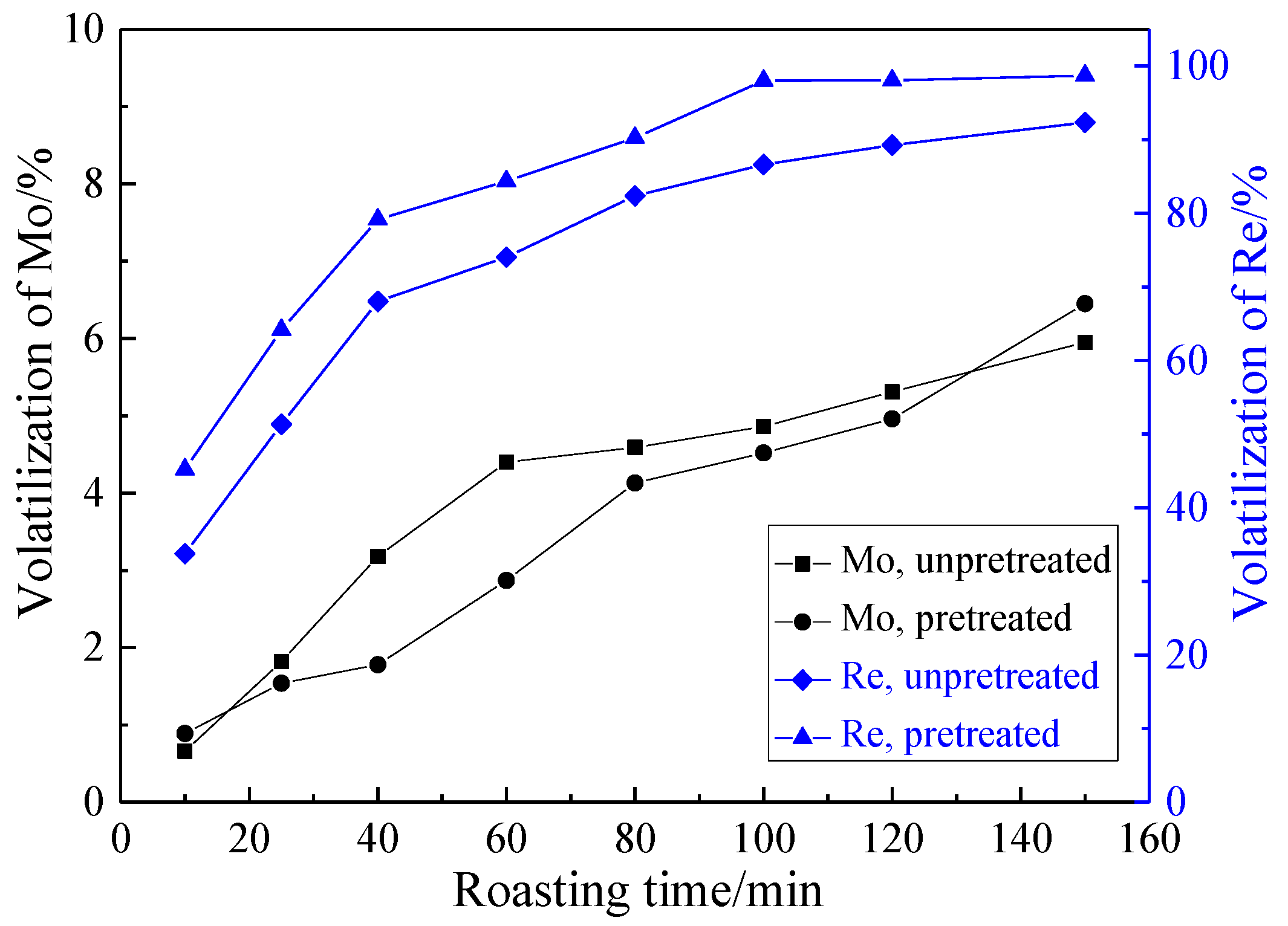
3.2.2. Effect of Roasting Time
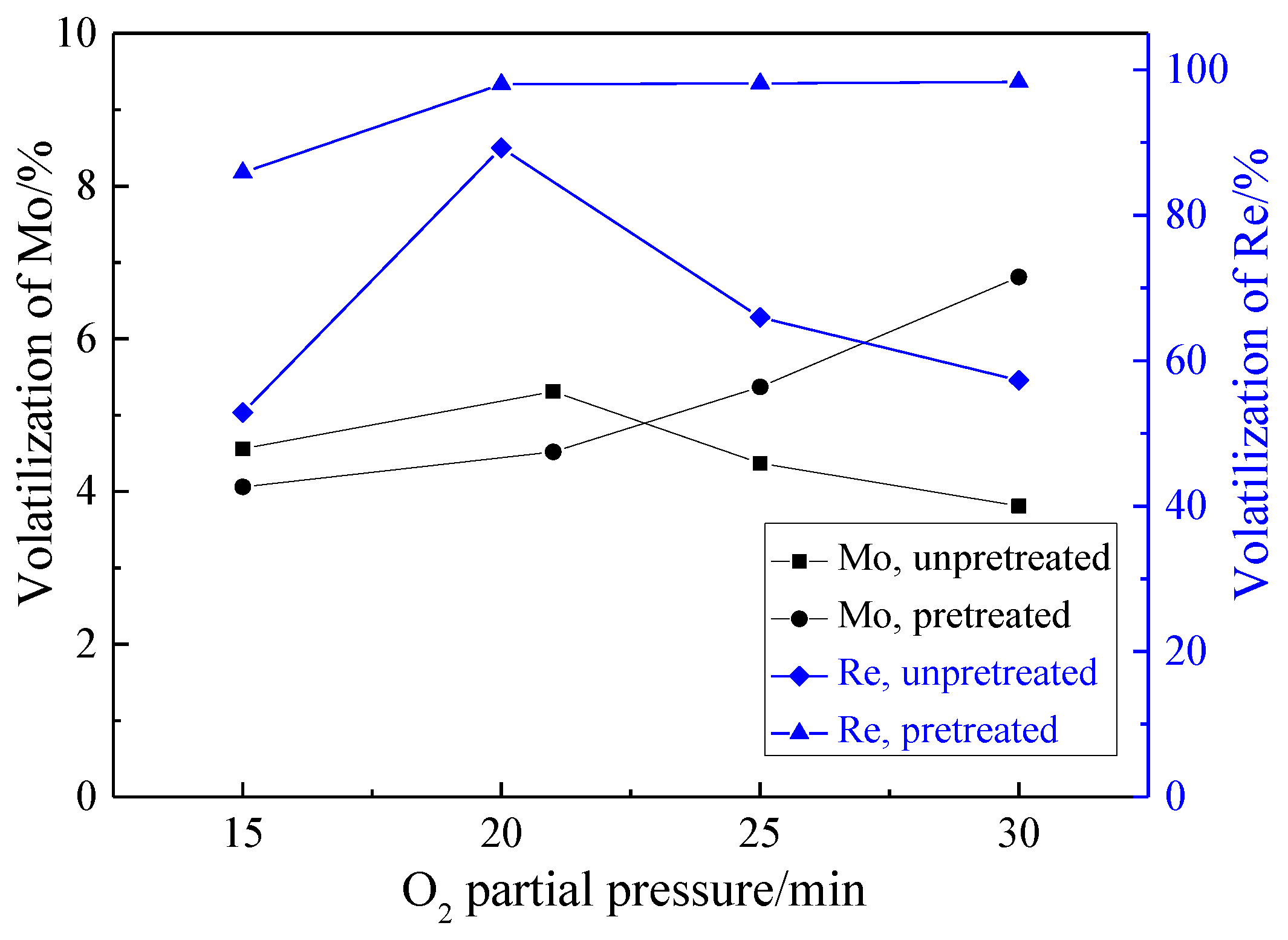
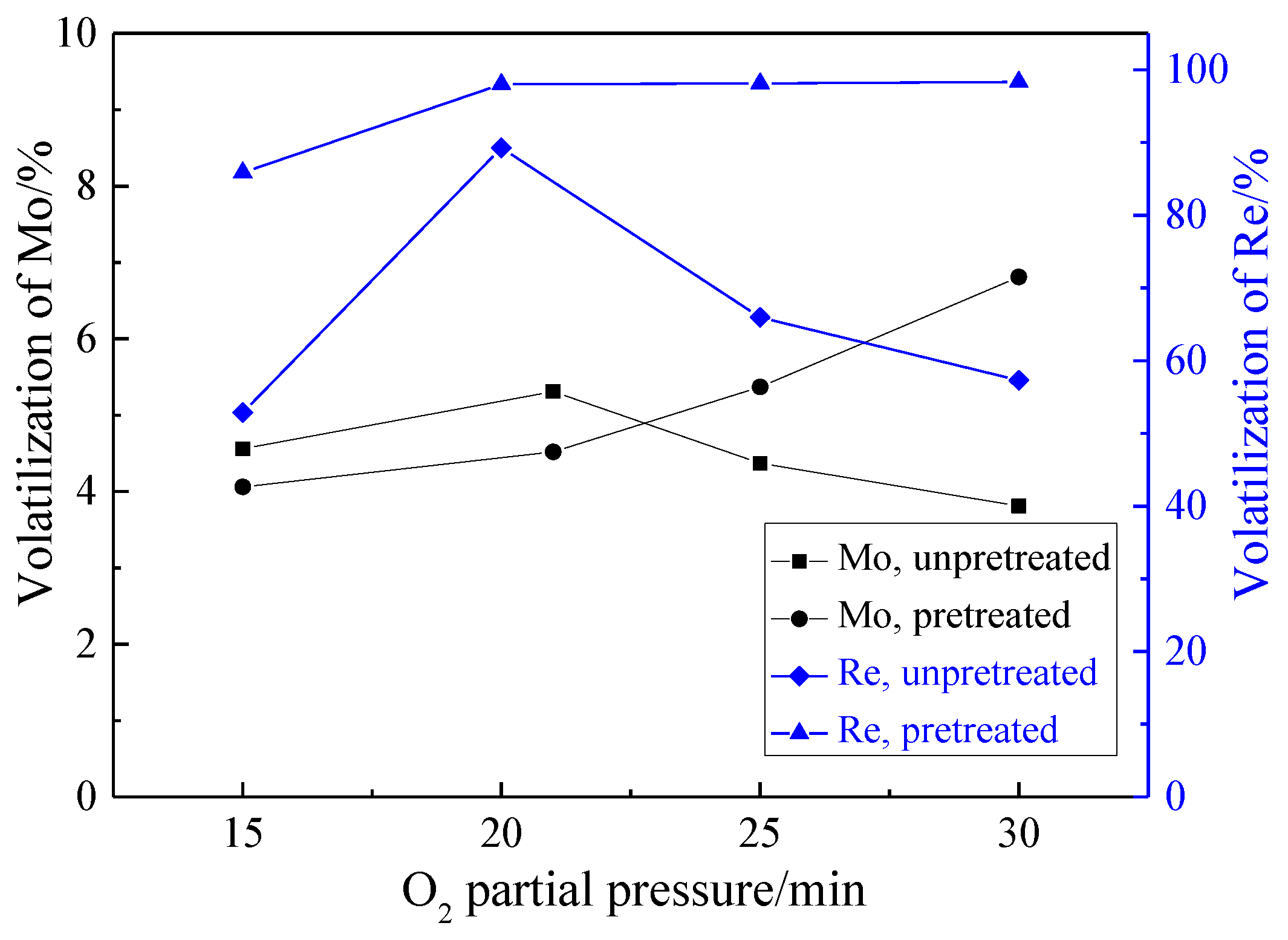
3.2.3. Effect of O2 Partial Pressure
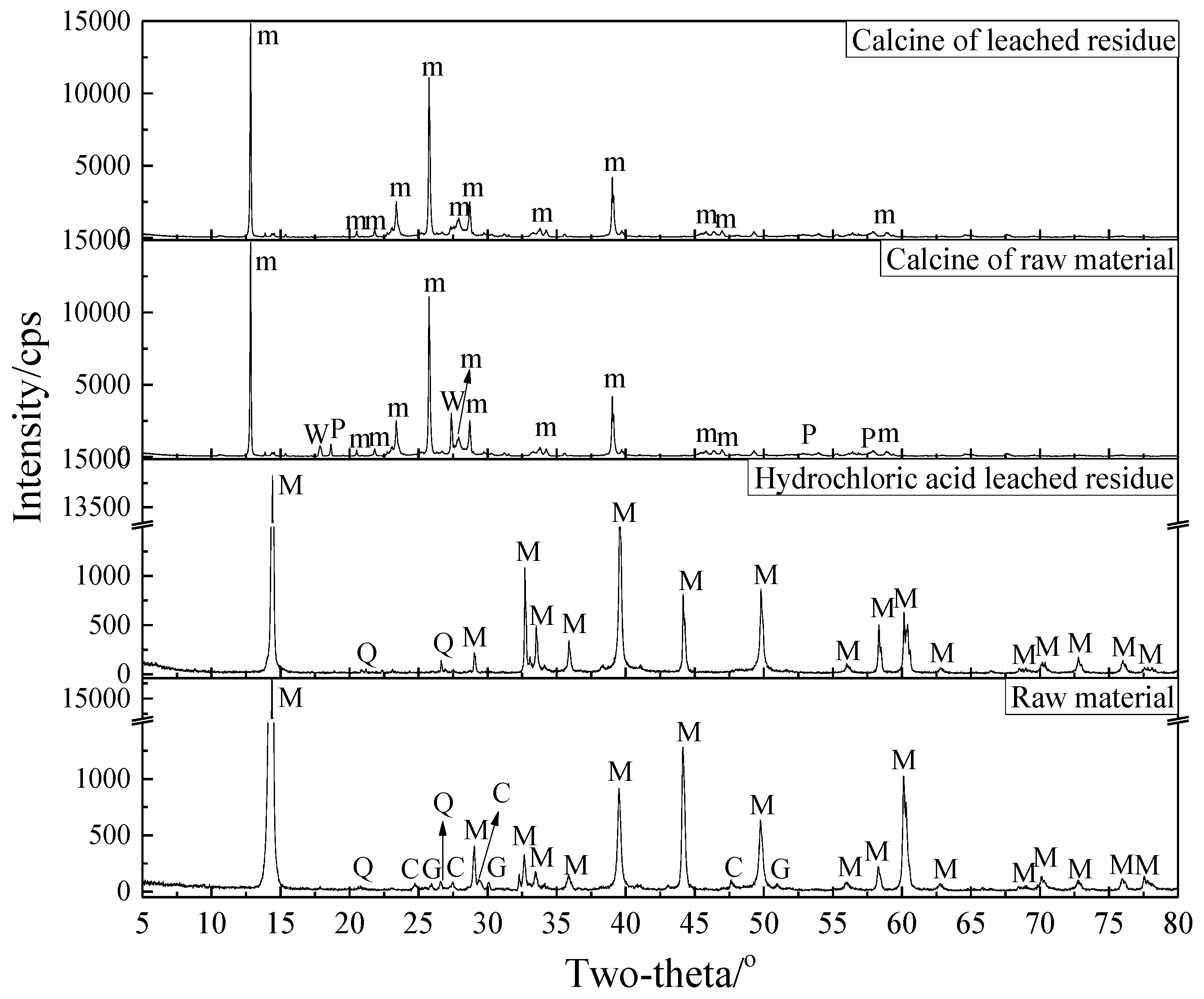
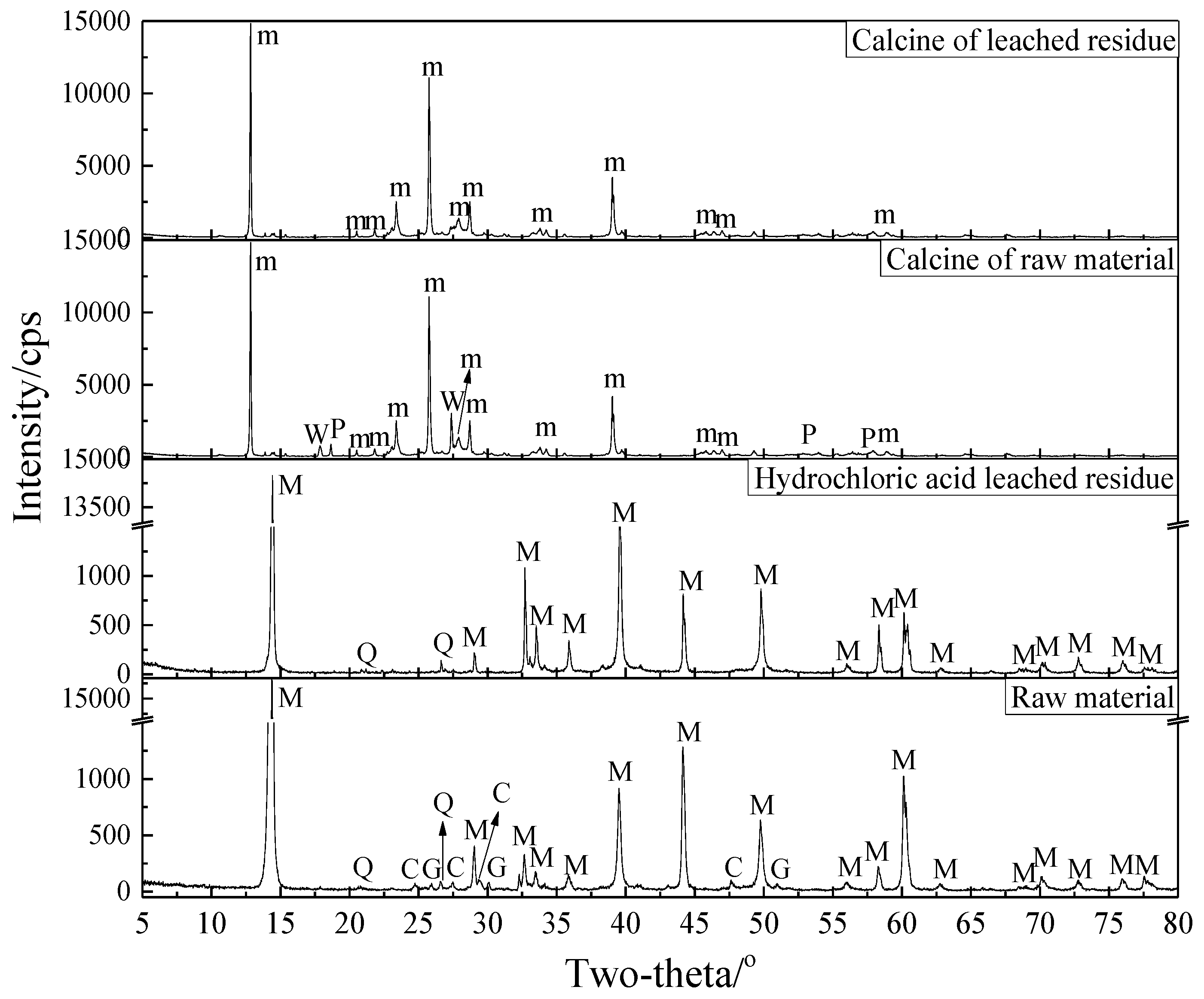
3.2.4. Phase Transformation in the Roasting Process
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hu, K.; Zhao, D.; Liu, J. Synthesis of nano-MoS2/bentonite composite and its application for removal of organic dye. Trans. Nonferr. Met. Soc. 2012, 22, 2484–2490. [Google Scholar] [CrossRef]
- U.S. Geological Survey. Minerals Commodity Summaries (2015) and Minerals Yearbook (2012). Available online: http://minerals.usgs.gov/minerals/pubs/commodity/rhenium/mcs-2012-rheni.pdf (accessed on 23 March 2015).
- Mccandless, T.; Ruiz, J.; Campbell, A. Rhenium behavior in molybdenite in hypogene and near-surface environments: Implications for Re-Os geochronometry. Geochim. Cosmochim. Acta 1993, 57, 889–905. [Google Scholar] [CrossRef]
- Xiang, T. Molybdenum Metallurgy; Central South University Press: Changsha, China, 2002; pp. 23–34. (In Chinese) [Google Scholar]
- Tarasov, A.; Besser, A.; Gedgagov, E. Integrated technology for processing rhenium-containing molybdenite concentrates to recover molybdenum and rhenium into commercial products. Miner. Process. Extr. Metall. Rev. 2001, 22, 509–517. [Google Scholar] [CrossRef]
- Shariat, M.; Hassani, M. Rhenium recovery from Sarcheshmeh molybdenite concentrate. J. Mater. Process. Technol. 1998, 74, 243–250. [Google Scholar] [CrossRef]
- Tripathy, P.; Rakhasia, R. Chemical processing of a low grade molybdenite concentrate to recover molybdenum. Trans. Inst. Min. Metall. Sect. C 2006, 115, 8–14. [Google Scholar] [CrossRef]
- Juneja, J.; Singh, S.; Bose, D. Investigations on the extraction of molybdenum and rhenium values from low grade molybdenite concentrate. Hydrometallurgy 1996, 41, 201–209. [Google Scholar] [CrossRef]
- Ketcham, V.; Hazen, W.; Coltrinari, E. Pressure oxidation process for the production of molybdenum trioxide from molybdenite. U.S. Patent US614988321, November 2000. [Google Scholar]
- Khoshnevisan, A.; Yoozbashizadeh, H.; Mozammel, M.; Sadrnezhaad, S. Kinetics of pressure oxidative leaching of molybdenite concentrate by nitric acid. Hydrometallurgy 2012, 111–112, 52–57. [Google Scholar] [CrossRef]
- Jiang, K.; Wang, Y.; Zou, X.; Zhang, L.; Liu, S. Extraction of molybdenum from molybdenite concentrates with hydrometallurgical processing. JOM 2012, 64, 1285–1289. [Google Scholar] [CrossRef]
- Joo, S.; Kim, Y.; Kang, J.; Yoon, H.; Kim, D.; Shin, S. Recovery of molybdenum and rhenium using selective precipitation method from molybdenite roasting dust in alkali leaching solution. Mater. Trans. 2012, 53, 2038–2042. [Google Scholar] [CrossRef]
- Cao, Z.; Zhong, H.; Liu, G.; Fu, J.; Wang, S.; Qiu, Y. Electric-oxidation kinetics of molybdenite concentrate in acidic NaCl solution. Can. J. Chem. Eng. 2009, 87, 939–944. [Google Scholar]
- Molaei, R.; Shariat, M.; Zakeri, A.; Boutorabi, M. Rhenium (VII) recovery via electro-oxidation process I. The influence of time, temperature, and pH. Electrochem. Solid. St. Lett. 2011, 14, E1–E3. [Google Scholar] [CrossRef]
- Askari Zamani, M.; Hiroyoshi, N.; Tsunekawa, M.; Vaghar, R.; Oliazadeh, M. Bioleaching of Sarcheshmeh molybdenite concentrate for extraction of rhenium. Hydrometallurgy 2005, 80, 23–31. [Google Scholar] [CrossRef]
- Olson, G.; Clark, T. Bioleaching of molybdenite. Hydrometallurgy 2008, 93, 10–15. [Google Scholar] [CrossRef]
- Peng, Z.; Hwang, J. Microwave-assisted metallurgy. Int. Mater. Rev. 2015, 60, 30–63. [Google Scholar] [CrossRef]
- Joo, S.; Kim, Y.; Kang, J.; Kumar, J.; Yoon, H.; Parhi, P. Recovery of rhenium and molybdenum from molybdenite roasting dust leaching solution by ion exchange resins. Mater. Trans. 2012, 53, 2034–2037. [Google Scholar] [CrossRef]
- Lan, X.; Liang, S.; Song, Y. Recovery of rhenium from molybdenite calcine by a resin-in-pulp process. Hydrometallurgy 2006, 82, 133–136. [Google Scholar] [CrossRef]
- Seo, S.; Choi, W.; Yang, T.; Kim, M.; Tran, T. Recovery of rhenium and molybdenum from a roaster fume scrubbing liquor by adsorption using activated carbon. Hydrometallurgy 2015, 129–130, 145–150. [Google Scholar] [CrossRef]
- Keshavarz Alamdari, E.; Darvishi, D.; Haghshenas, D.; Yousefi, N.; Sadrnezhaad, S. Separation of Re and Mo from roasting-dust leach-liquor using solvent extraction technique by TBP. Sep. Purif. Technol. 2012, 86, 143–148. [Google Scholar] [CrossRef]
- Marin, T.; Utigard, T.; Hernandez, C. Roasting kinetics of molybdenite concentrates. Can. Metall. Q. 2009, 48, 73–80. [Google Scholar] [CrossRef]
- Mchugh, L.; Balliett, R.; Mozolic, J. The sulfide ore looping oxidation process: An alternative to current roasting and smelting practice. JOM 2008, 60, 84–87. [Google Scholar] [CrossRef]
- Kim, B.; Lee, H.; Choi, Y.; Kim, S. Kinetics of the oxidative roasting of low grade Mongolian molybdenite concentrate. Mater. Trans. 2009, 50, 2669–2674. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Dang, J.; Chou, K. Oxidation roasting of molybdenite concentrate. Trans. Nonferr. Met. Soc. 2015, 25, 4167–4174. [Google Scholar] [CrossRef]
- Shigegaki, Y.; Basu, S.; Wakihara, M.; Taniguchi, M. Thermal analysis and kinetics of oxidation of molybdenum sulfides. J. Therm. Anal. Calorim. 1988, 34, 1427–1440. [Google Scholar] [CrossRef]
- Utigard, T. Oxidation mechanism of molybdenite concentrate. Metall. Mater. Trans. B 2009, 40, 490–496. [Google Scholar] [CrossRef]
- Ma, G.; Lei, N.; Wang, Z.; Wang, W.; Feng, B. Research on reducing the lead content of high-lead molybdenite concentrates. China Molybd. Ind. 2015, 39, 23–25. (In Chinese) [Google Scholar]
- Gupta, C. Extractive Metallurgy of Molybdenum; CRC Press: Boca Raton, FL, USA, 1992; pp. 156–157. [Google Scholar]
- Holdich, R.; Lawson, G. The solubility of aqueous lead chloride solutions. Hydrometallurgy 1987, 19, 199–208. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mo | Re | Fe | SiO2 | Al2O3 | CaO | MgO | WO3 | Cu | Pb | S |
|---|---|---|---|---|---|---|---|---|---|---|
| 43.55 | 321 g/t | 1.39 | 4.02 | 0.18 | 3.27 | 0.48 | 1.66 | 0.03 | 4.52 | 32.46 |
| Component | Mo | Re | Fe | SiO2 | Al2O3 | CaO | MgO | WO3 | Cu | Pb | S |
|---|---|---|---|---|---|---|---|---|---|---|---|
| wt. % | 50.25 | 363 g/t | 1.07 | 4.72 | 0.53 | 0.10 | 0.18 | 1.76 | 0.02 | 0.34 | 33.50 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; You, Z.; Sun, H.; Sun, R.; Peng, Z.; Zhang, Y.; Jiang, T. Separation of Rhenium from Lead-Rich Molybdenite Concentrate via Hydrochloric Acid Leaching Followed by Oxidative Roasting. Metals 2016, 6, 282. https://doi.org/10.3390/met6110282
Li G, You Z, Sun H, Sun R, Peng Z, Zhang Y, Jiang T. Separation of Rhenium from Lead-Rich Molybdenite Concentrate via Hydrochloric Acid Leaching Followed by Oxidative Roasting. Metals. 2016; 6(11):282. https://doi.org/10.3390/met6110282
Chicago/Turabian StyleLi, Guanghui, Zhixiong You, Hu Sun, Rong Sun, Zhiwei Peng, Yuanbo Zhang, and Tao Jiang. 2016. "Separation of Rhenium from Lead-Rich Molybdenite Concentrate via Hydrochloric Acid Leaching Followed by Oxidative Roasting" Metals 6, no. 11: 282. https://doi.org/10.3390/met6110282
APA StyleLi, G., You, Z., Sun, H., Sun, R., Peng, Z., Zhang, Y., & Jiang, T. (2016). Separation of Rhenium from Lead-Rich Molybdenite Concentrate via Hydrochloric Acid Leaching Followed by Oxidative Roasting. Metals, 6(11), 282. https://doi.org/10.3390/met6110282