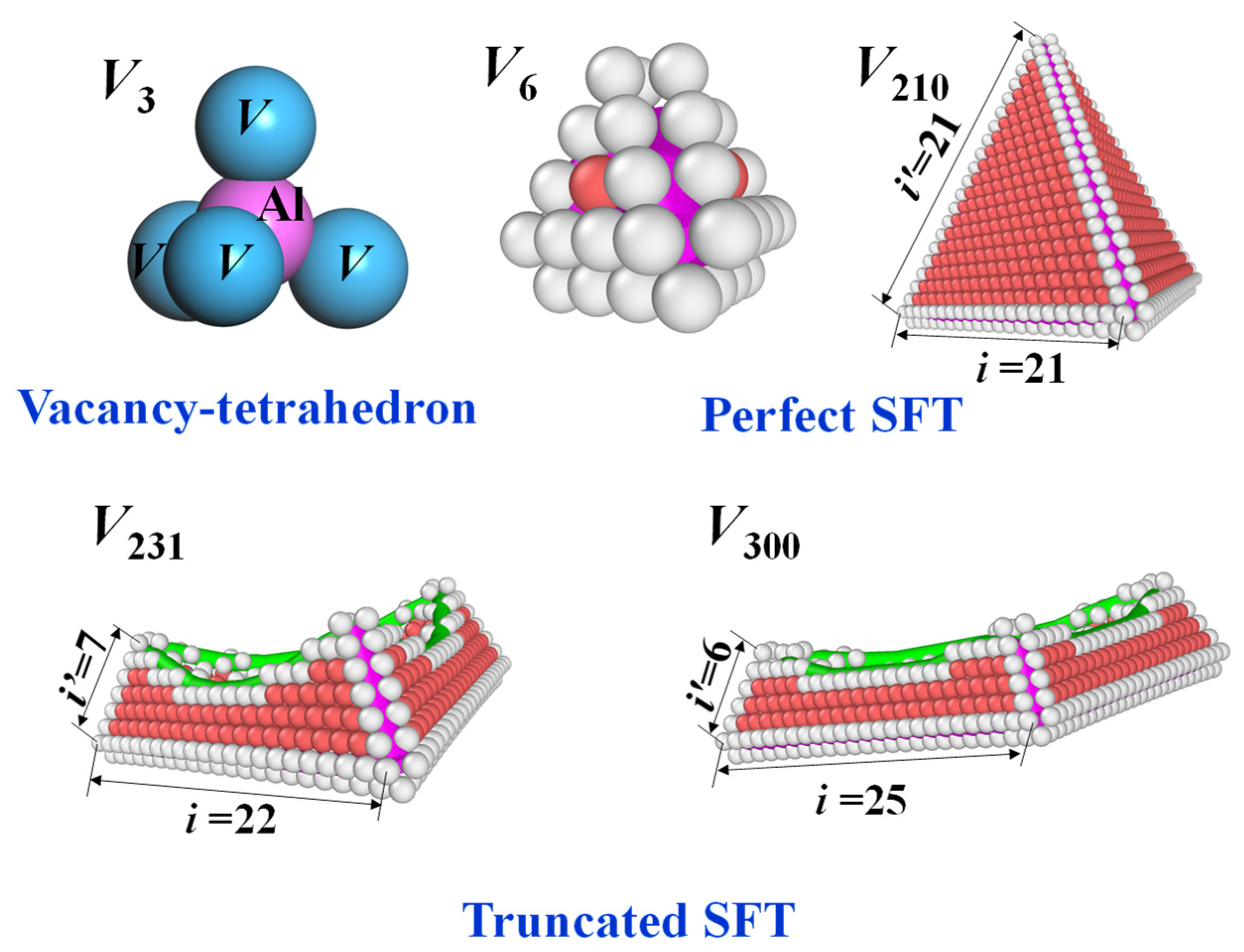
3.1. Structures of SFT
The perfect SFT adopts a highly symmetric tetrahedral morphology, comprising six 1/6<110> stair-rod dislocations along its edges and four intrinsic stacking faults confined to the {111} planes (see
Figure 1). The size of such structures is quantized, corresponding to specific “magic numbers” of vacancies defined by
, where
is an integer starting from 3. This yields vacancy counts of 3, 6, 10, 15, 21, 28, and so forth. Notably, the case of
, which nominally corresponds to three vacancies, does not result in a perfect SFT. Instead, it forms a tetrahedron consisting of four nearest-neighbor vacancies enclosing an interstitial aluminum atom. This configuration is usually denoted as a “vacancy-tetrahedron” (see
Figure 1). The smallest structure that qualifies as a perfect SFT appears at
, corresponding to
(see
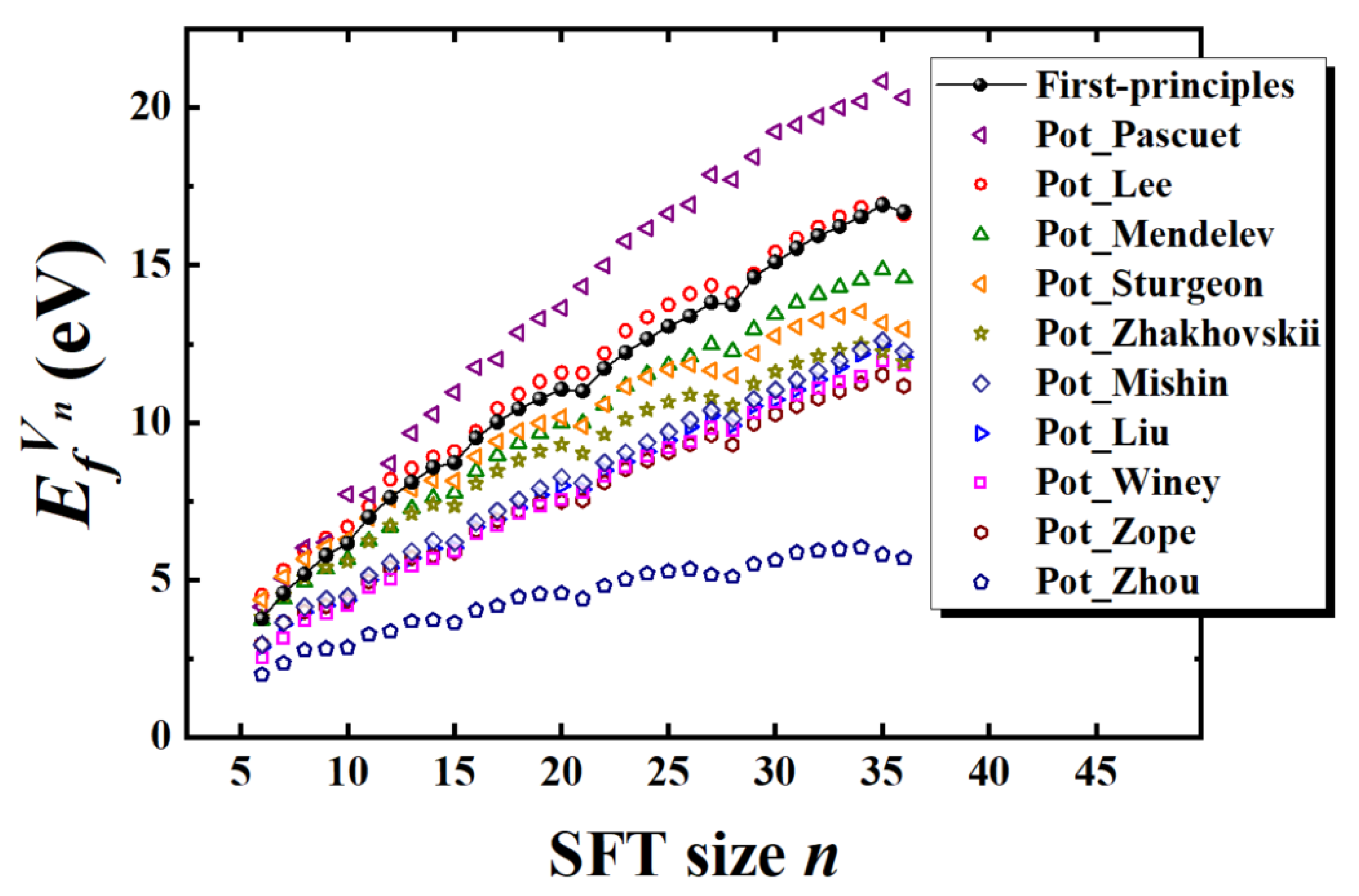
Figure 1). Perfect SFT configurations can be systematically generated via MS simulations by introducing equilateral triangular vacancy plates into the (111) planes of the FCC matrix and relaxing the structure, following the Silcox–Hirsch mechanism. Among the ten empirical interatomic potentials evaluated, Pot_Zhakhovskii proved most effective in stabilizing perfect SFTs, supporting structures containing up to 780 vacancies. In contrast, other potentials failed to preserve the ideal tetrahedral symmetry at larger sizes—typically beyond 200 to 300 vacancies—resulting instead in truncated configurations, that is, “truncated SFT” (see
Figure 1).
The truncated SFT is characterized by the absence of one vertex of the tetrahedron. Specifically, it terminates at the junction of three Shockley partial dislocations, resulting in a geometry where the top facet is re-entrant due to the high intrinsic stacking fault energy of aluminum (see
Figure 1). As additional vacancies are incorporated, the structure undergoes further distortion, becoming increasingly compressed and ultimately transforming into a dissociated Frank-type dislocation loop—a transition consistent with prior theoretical predictions. Taking the Pot_Lee potential as a representative example, the formation of truncated SFTs begins at approximately 231 vacancies. The structural height of the truncated SFT, denoted as
in
Figure 1, decreases rapidly with increasing vacancy number. As shown in
Figure 2,
converges to a value of 6 by
, beyond which it remains invariant despite further increases in vacancy count. These results are in agreement with those reported by Zhang et al. [
9].
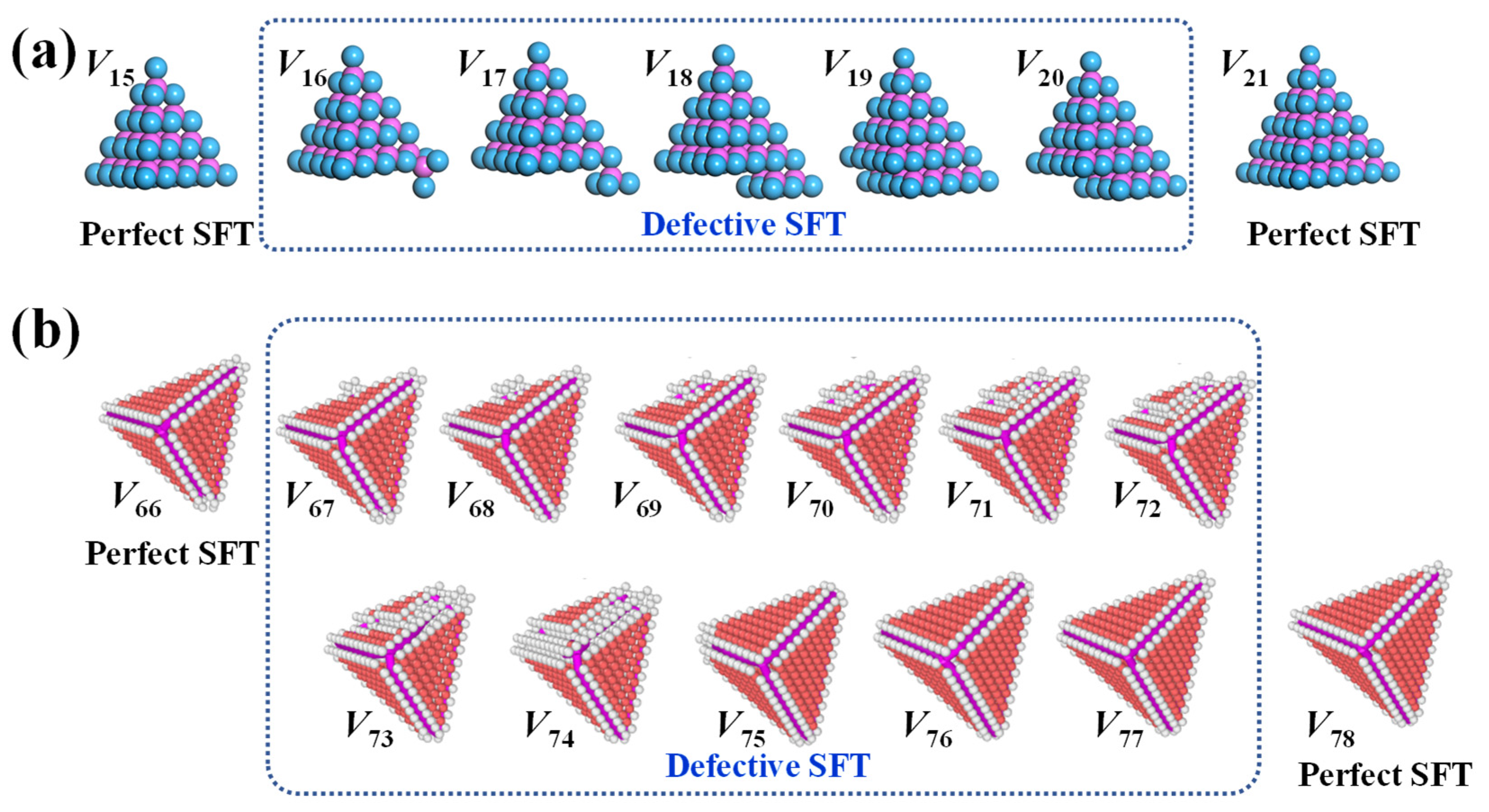
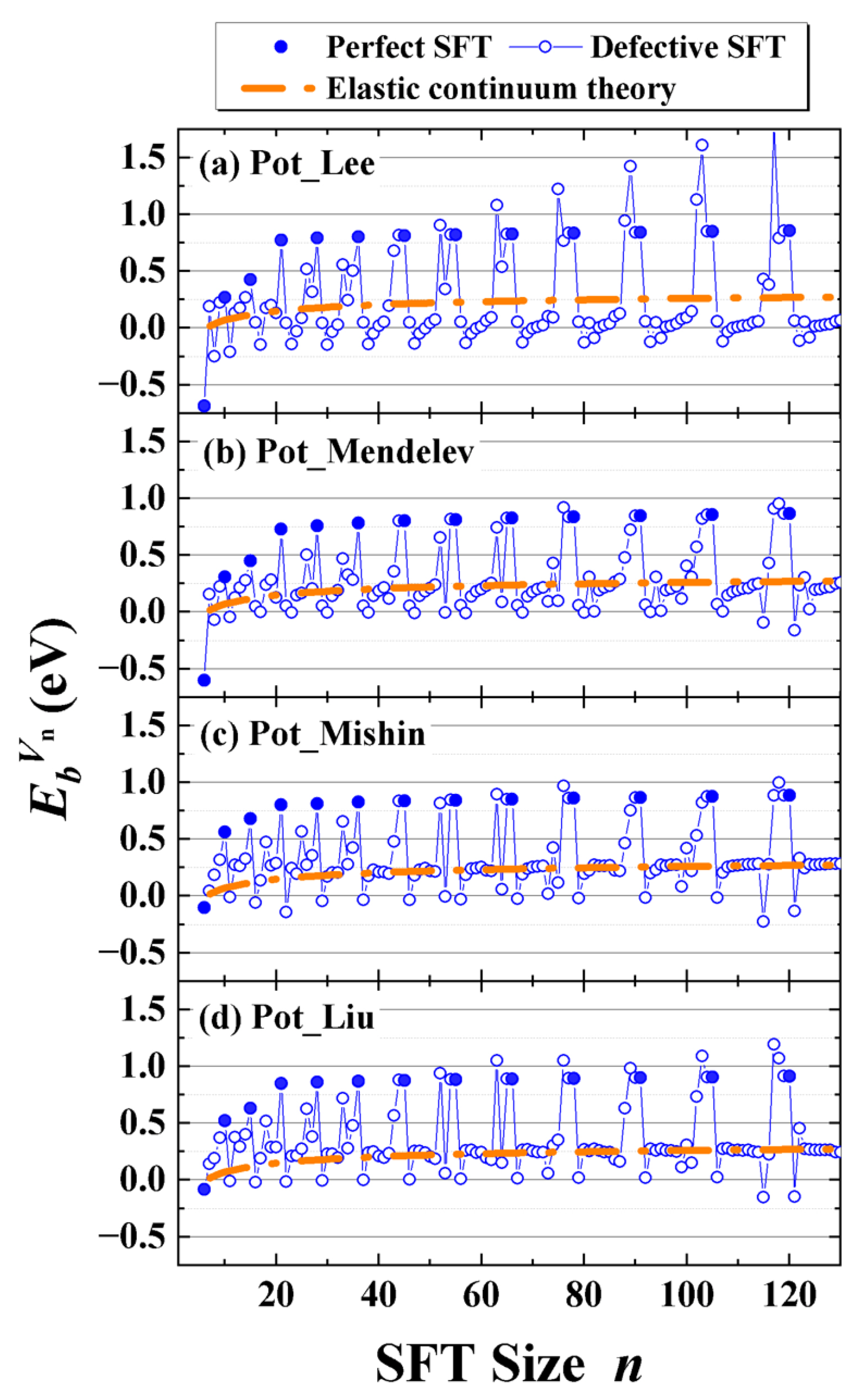
While the structural characteristics of perfect and truncated SFTs have been extensively investigated, our simulations reveal a third, previously underexplored variant: the defective SFT. These configurations emerge between two perfect SFTs. Defective SFTs can also be obtained via the Silcox–Hirsch mechanism, by introducing additional vacancies at lattice sites near the perimeter of an equilateral triangular vacancy plate embedded in the (111) plane. Upon structural relaxation through MS simulations, these perturbed vacancy plates evolve into defective SFTs. To systematically identify energetically favorable defective SFT configurations, we performed short-time MD simulations at representative temperatures (300 K and 600 K), using the aforementioned triangular vacancy plates as initial conditions. A series of intermediate structures was extracted from the MD trajectories and subsequently subjected to energy minimization via MS relaxation. The configuration yielding the lowest energy at each vacancy number was regarded as the most stable defective SFT and selected for detailed analysis. These optimized structures provided the basis for calculating the formation and binding energies, thereby enabling a thermodynamic assessment of the relative stability and growth behavior of defective SFT in aluminum.
Figure 3 presents two representative series of defective SFTs identified in the vacancy ranges between perfect SFTs with magic number counts of
-
and
-
. These defective configurations consistently preserve the fundamental tetrahedral geometry characteristic of the preceding smaller perfect SFT, yet exhibit incompleteness relative to the next higher-order magic-number structure. Detailed analysis reveals that the formation of these intermediate structures begins with the adsorption of vacancies either at the tetrahedral corners (e.g.,
) or at the midpoints along stair-rod dislocation edges (e.g.,
). These initial additions give rise to local vacancy-tetrahedron configuration. As more vacancies are incorporated, they preferentially accumulate along one {111} facet of the tetrahedron, initiating the nucleation and growth of an embryonic stacking fault (e.g.,
to
and
to
). This evolution continues incrementally until the total vacancy count approaches the next magic number. Strikingly, when the vacancy number falls within three to four of the next perfect SFT configuration, a pronounced structural transition occurs. As shown in configurations such as
to
, the defective SFT often manifests incomplete vertices at the corners of the tetrahedron, in comparison to the fully enclosed form of the subsequent perfect SFT. These observations illustrate the progressive evolution of defective SFTs as they transition toward the next perfect magic-number configuration.
3.5. Formation and Growth of SFTs via Vacancy Aggregation Mechanism
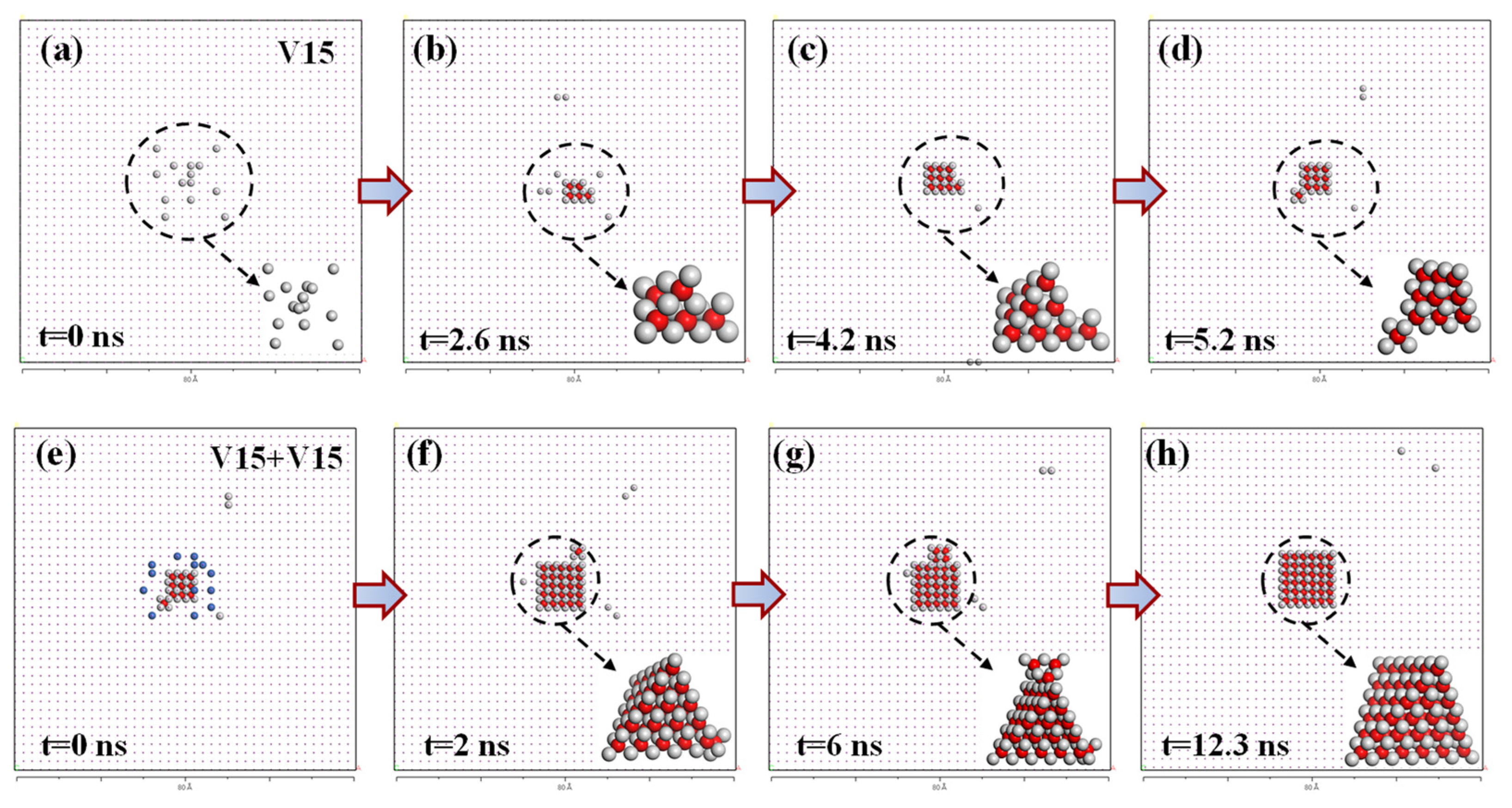
The SFT growth process described above is inferred solely from thermodynamic data—namely, the evolution of stable configurations and binding energies as a function of cluster size—indicating a thermodynamically favorable trend for incremental growth via vacancy capture. To further elucidate the formation and growth of SFTs via vacancy aggregation, we performed MD simulations using the Pot_Mendelev potential. The simulations were conducted at 600 K within a 20 × 20 × 20 supercell. At the onset, 15 vacancies were randomly introduced near the center of the simulation box (see
Figure 8a). Over time, these vacancies began to cluster, and at 2.6 ns, a defective SFT comprising eight vacancies emerged (see
Figure 8b). This defective SFT structure formed via the attachment of an additional vacancy to the smallest perfect SFT
through a vacancy-tetrahedron (see the inset at the lower right of
Figure 8b). Prior to the formation of the SFT
, a transient vacancy-tetrahedron was briefly observed; however, this intermediate configuration proved unstable and rapidly dissociated. The emergence of a stable SFT structure began with the formation of
, which subsequently grew through the sequential absorption of nearby vacancies. It is important to note that certain stable vacancy cluster configurations (such as
, which will be discussed later) may exist but were not observed during this sampling. This absence is likely due to the limitations inherent in the sampling process rather than the nonexistence of such structures. By 4.2 ns, the structure evolved into a larger defective SFT
, comprising a perfect SFT
and an adjacent vacancy-tetrahedron located at one of its vertices (see
Figure 8c). Continued vacancy capture led to the development of a defective SFT
by 5.2 ns, leaving only three isolated vacancies remaining in the system at that time (see
Figure 8d).
To investigate further growth behavior, 15 additional vacancies were introduced near the SFT
structure (see
Figure 8e, where the newly added vacancies are marked by small blue spheres), and a new MD simulation was initiated. At 2 ns into this subsequent simulation, a portion of these new vacancies had been captured by pre-existing SFT segments, leading to the formation of a 22-vacancy SFT. Continued evolution resulted in a 25-vacancy SFT by 2.6 ns. Structural comparison between the configurations at 2 ns and 6 ns revealed that, apart from additional apex vacancies, new tetrahedral substructures were observed to nucleate and expand from the midpoint along the edges of the pre-existing SFT. This observation aligns with the static predictions described earlier, which suggest that SFT growth preferentially initiates at the corners or edge midpoints of the existing tetrahedral structure. By 12.3 ns, this defective SFT had further matured into a perfect SFT
configuration through absorption of nearly all remaining vacancies—only two vacancies remained unincorporated.
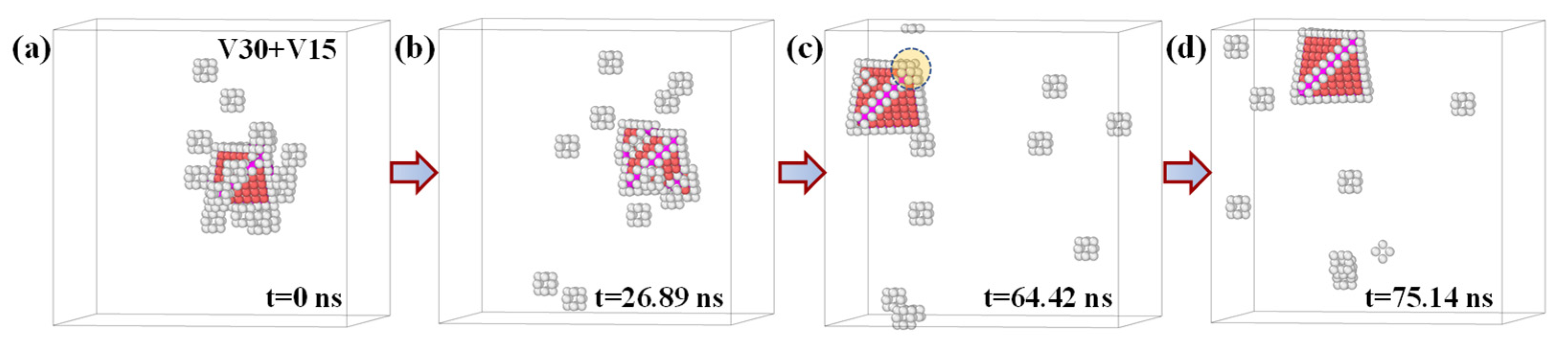
These MD simulations provide direct atomistic evidence that, in aluminum, SFT can indeed nucleate and grow purely via vacancy agglomeration. Importantly, the entire formation and growth process occurred via vacancy diffusion and aggregation. To further investigate the continued evolution of SFTs via vacancy-driven mechanisms, we repeated the aforementioned “vacancy introduction–growth” MD simulation protocol. Specifically, we used the final configuration from the previous MD simulation containing 30 vacancies as the initial structure and introduced an additional 15 vacancies. The subsequent evolution of the SFT structure and surrounding vacancies is illustrated in
Figure 9. At t = 26.89 ns, the SFT exhibited growth through vacancy capture at one of its corners, forming an irregular defective region. Concurrently, an embryonic stacking fault emerged on one of the SFT facets, signaling a secondary nucleation site for further structural development. By t = 64.42 ns, a nearly complete SFT comprising 35 vacancies had formed, characterized by a single defect located at one of its vertices. This minor imperfection was resolved by t = 75.14 ns, when the capture of an additional vacancy transformed the structure into a perfect SFT
. These results further strengthen the atomistic evidence that SFTs in aluminum can grow via successive vacancy absorption. Notably, the observed growth pathway involves both vertex- and facet-mediated vacancy capture, consistent with our earlier thermodynamic analysis that predicted preferential growth at corners and along facet edges. In particular, the simultaneous vacancy incorporation at both a vertex and a facet observed at t = 26.89 ns highlights the cooperative and spatially distributed nature of SFT maturation, suggesting that its growth is governed by a more complex and synergistic mechanism than previously assumed.
Figure 9 presents four representative snapshots of the evolving system, all rendered from a consistent viewpoint to clearly illustrate the time-resolved morphological evolution of the SFT and the surrounding vacancy distribution. Notably, beyond the progressive capture and redistribution of vacancies, the spatial position of the SFT itself exhibits significant displacement throughout the simulation. Initially located near the center of the supercell at t = 0 ns, the SFT migrates toward the upper left region of the simulation box by t = 75.14 ns. This substantial relocation offers compelling evidence that, under the present thermal conditions, the SFT possesses notable mobility. This observation stands in stark contrast to the conventional understanding of SFTs as immobile defects. Traditionally, SFTs are considered sessile due to their bounding stair-rod dislocations, whose glide plane is not a compact {111} plane. The finding here directly challenges this long-standing view.
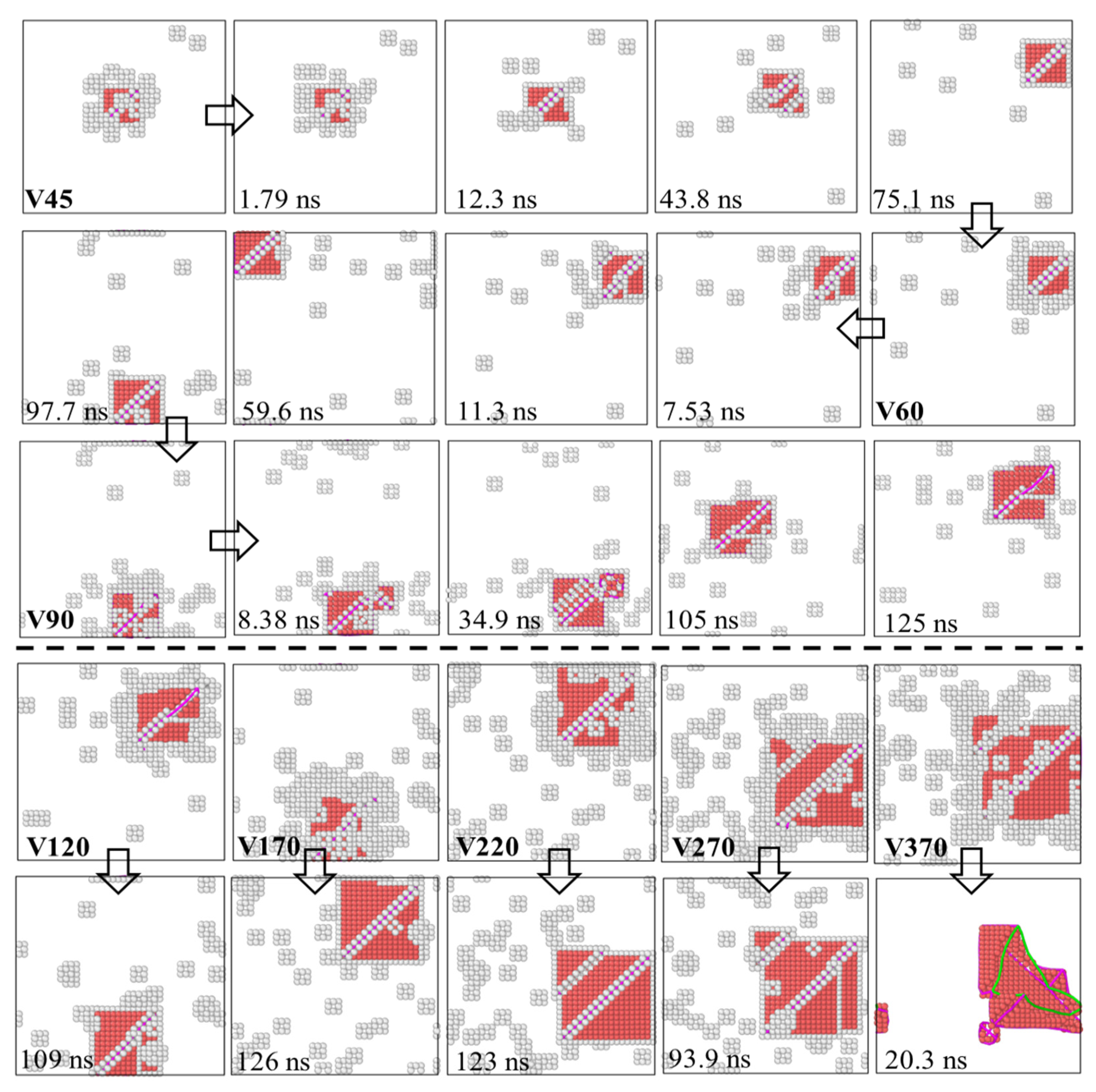
To further investigate the growth dynamics and structural evolution of SFTs driven by vacancy absorption, we systematically extended our MD simulation strategy based on a vacancy-introduction and growth approach. The initial system, denoted as
, consisted of a perfect SFT
and nine isolated vacancies. Building upon this configuration, additional vacancies were introduced stepwise to construct increasingly complex systems: 15 vacancies were first added to yield the
system, followed by successive additions of 30 vacancies to form the
and
systems. Subsequently, three consecutive batches of 50 vacancies each were introduced, generating the
,
, and
structures. Finally, an additional 100 vacancies were incorporated into the
system to produce the
configuration. Representative structural snapshots along this simulation trajectory are shown in
Figure 10, with all structural snapshots visualized from an identical orientation, enabling direct comparison of the evolving SFT geometry and the spatiotemporal distribution of vacancies throughout the simulation. The results reveal that the SFT continuously grow by absorbing vacancies, and exhibit mobility during the process. As the simulations progressed to the
system, we observed that the conventional SFT architecture became unstable, transforming into a truncated SFT. Further vacancy introduction—up to approximately 700 vacancies—resulted in the destabilization of the truncated SFT itself, which eventually evolved into a dislocation loop structure.
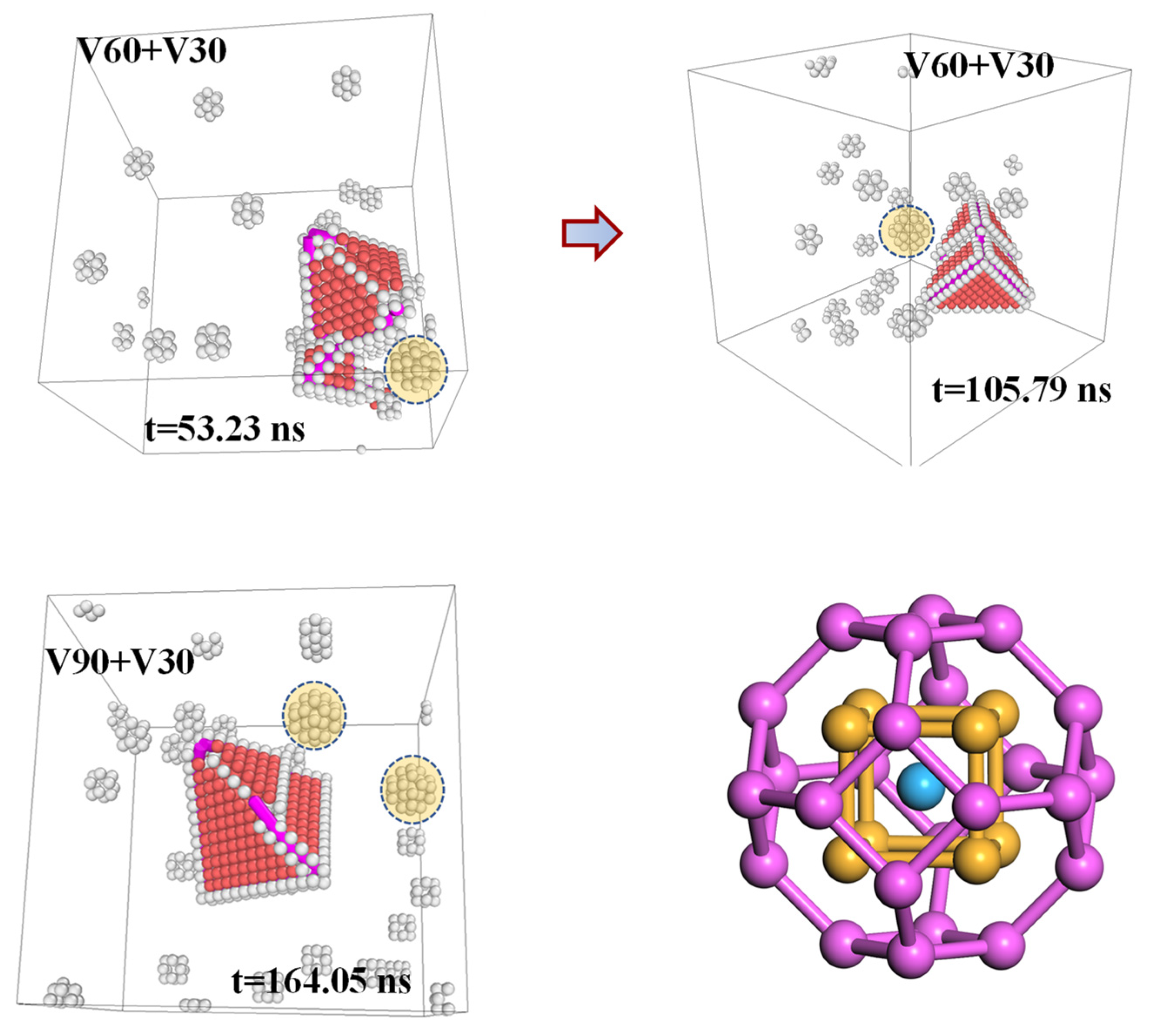
Notably, throughout the simulations, we frequently observed compact vacancy clusters in addition to isolated vacancies. In multiple snapshots—such as those from the
simulation at t = 53.23 ns and t = 105.79 ns, and the
simulation at t = 164.05 ns (
Figure 11)—truncated octahedral clusters composed of aluminum atoms were identified, featuring a core of nine atoms arranged in a body-centered cubic (BCC) configuration. This structure corresponds to the highly stable vacancy cluster
previously confirmed by previous studies and is widely regarded as a key precursor in the nucleation and growth of vacancy aggregates in aluminum [
7,
35]. Furthermore, an intriguing phenomenon was observed in the
simulation. Starting from a structure containing a perfect SFT
and 10 isolated vacancies, the addition of 30 new vacancies followed by MD simulation led to localized vacancy absorption at one corner of the original SFT, triggering the formation of a new, smaller SFT (see
Figure 11, t = 53.23 ns). As the simulation progressed, this nascent SFT evolved to display early-stage faulting across two of its facets (
Figure 11, t = 105.79 ns), further underscoring the complexity and cooperativity of SFT growth mechanisms.
These MD simulations presented above offer compelling atomistic evidence that SFTs in aluminum can nucleate spontaneously through vacancy agglomeration, without the involvement of dislocation mechanisms. The formation process initiates from the coalescence of small vacancy clusters and proceeds via the progressive absorption of nearby vacancies, ultimately leading to the emergence of a stable SFT. Beyond their ability to grow, the simulations also reveal that SFTs possess notable mobility—an often-overlooked characteristic with profound implications for their structural evolution. The observed mobility of SFTs plays a critical role in their transformation dynamics. On the one hand, mobile SFTs are more effective at capturing surrounding vacancies and can even merge with neighboring SFTs, facilitating cooperative growth. On the other hand, as vacancy accumulation continues, larger SFTs exhibit structural instability and eventually evolve into vacancy-type dislocation loops. This finding redefines SFTs not as terminal defect states but rather as metastable intermediates in a continuous vacancy-driven transformation pathway. This mechanistic insight provides a rational explanation for several long-standing experimental observations regarding SFTs in aluminum [
5]. In particular, experimental studies consistently report that SFTs in aluminum are typically very small—on the order of ~2 nm—and are predominantly observed under low-temperature conditions [
5]. Such behavior is likely a direct consequence of their intrinsic mobility: mobile SFTs readily coalesce or transform into more complex defect structures, such as dislocation loops, rendering them difficult to detect. In contrast, under cryogenic conditions, vacancy diffusion and SFT migration are suppressed, allowing these structures to persist in an isolated, observable form. Thus, the spontaneous formation and high mobility of SFTs are not only fundamental to their growth and transformation but also directly impact their detectability and morphology in experimental systems.
3.6. Comparison with Literature on SFT Formation Energies and Dynamics in FCC Metals
The vacancy formation energies predicted by the ten empirical potentials evaluated in this study range from 0.66 to 0.89 eV, which are in good agreement with both experimental values (0.62–0.72 eV) and DFT calculations (0.5–0.82 eV) reported in the literature [
7]. This agreement supports the reliability of these potentials in capturing basic defect energetics. To further assess the validity of our proposed formulation for stacking fault tetrahedron (SFT) formation energy, we applied Equation (4) to data extracted from a recent study by Landeiro Dos Reis et al. on aluminum [
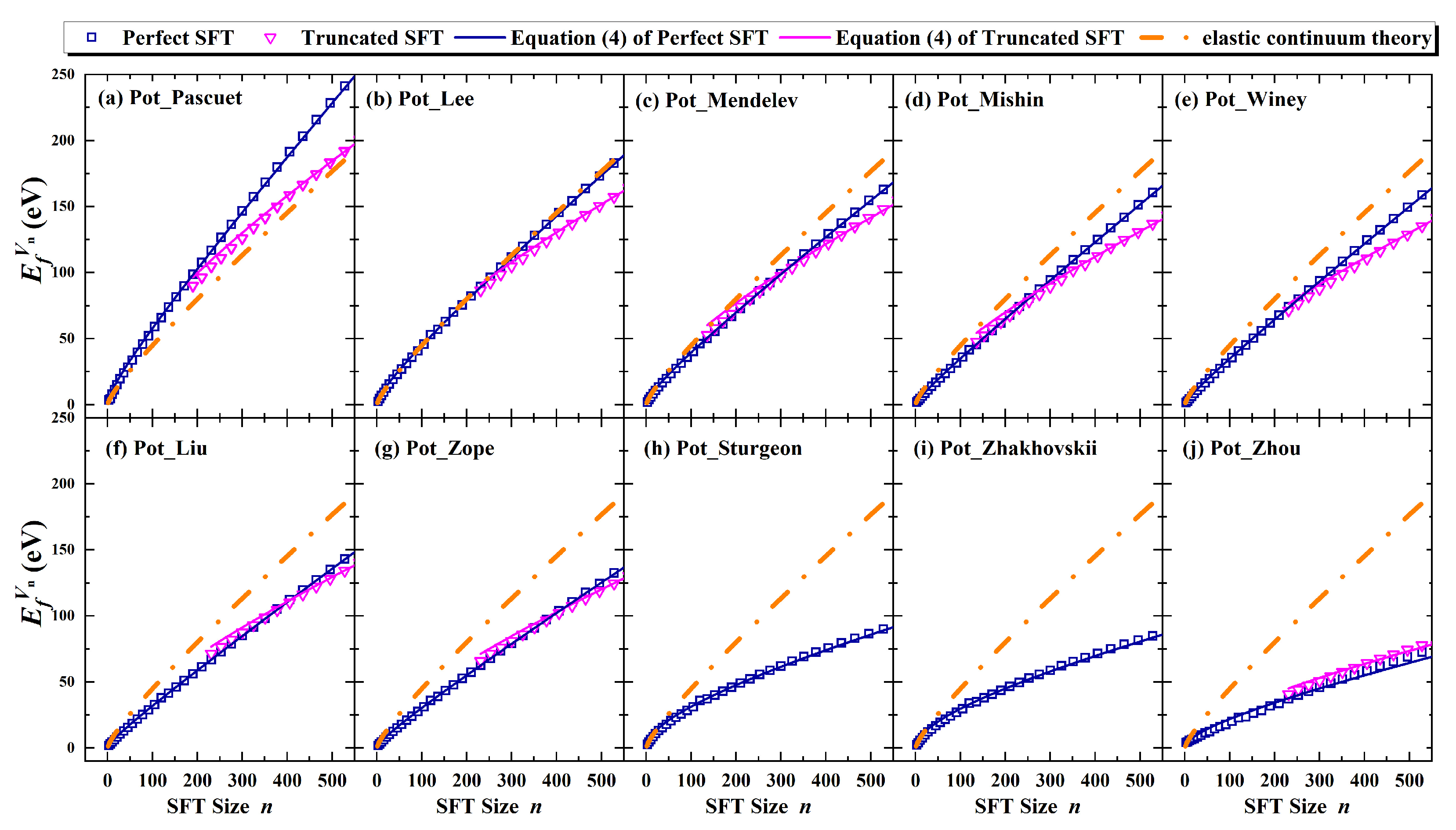
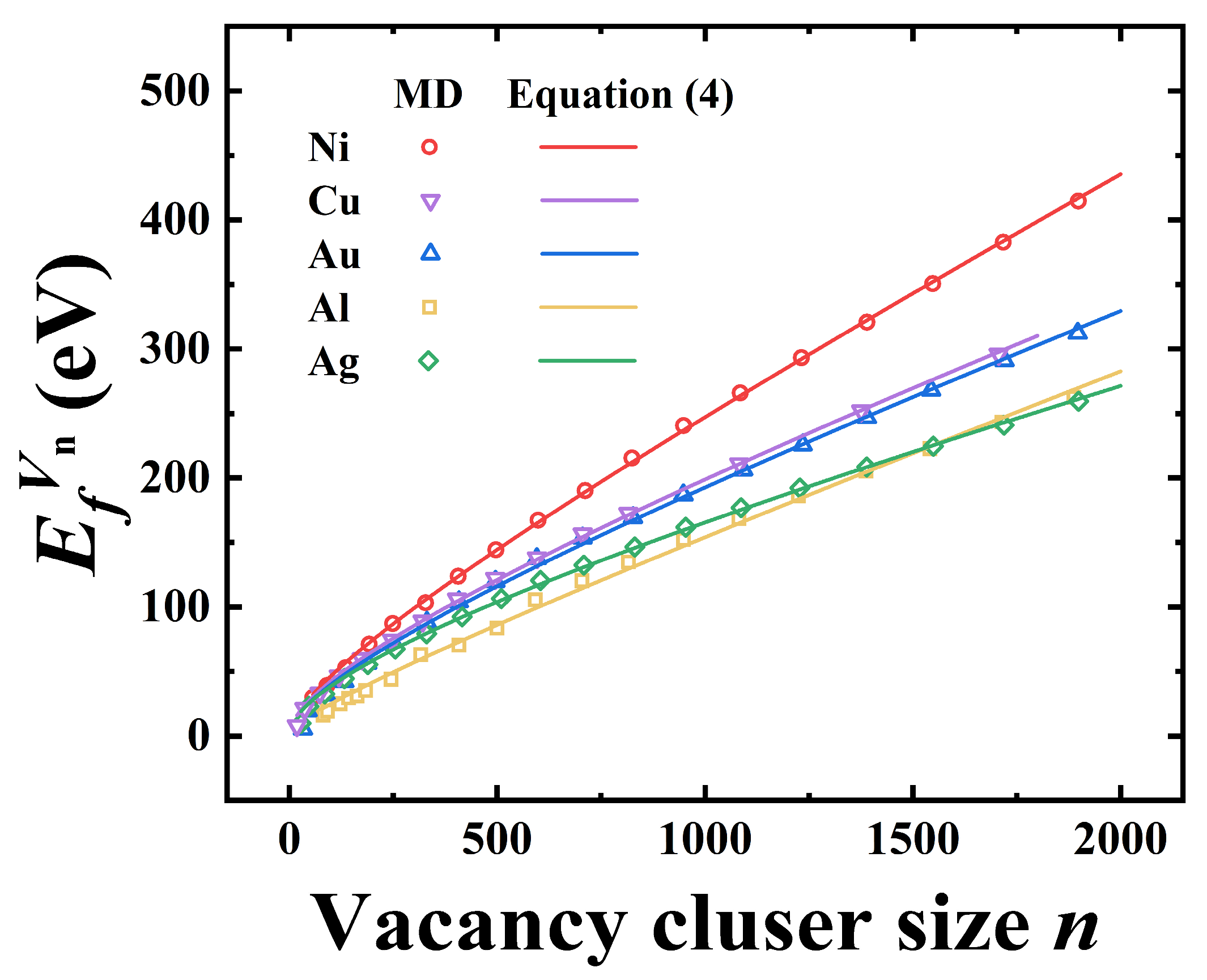
12]. Our results show that Equation (4) effectively describes the size dependence of SFT formation energies, indicating the robustness of our model.
Moreover, the general applicability of Equation (4) was tested by extending it to other FCC metals, including Ni, Cu, Au, and Ag, using additional data provided by Landeiro Dos Reis et al. [
12]. We found that the equation also captures the SFT formation energy trends in these metals, demonstrating the model’s versatility. These results are summarized in
Figure 12 and further strengthen the validity of the proposed formulation across a range of FCC metals.
In terms of the dynamic behavior of SFTs, MD simulations by Martínez and Uberuaga in Cu revealed that small, imperfect SFTs exhibit high mobility, allowing them to migrate and coalesce into larger, more stable structures [
36]. This diffusion-assisted growth mechanism suggests that SFT formation can occur through vacancy mobility, rather than being limited to direct cascade effects. Complementary experimental work by Aidhy et al. in Ni supports this view, showing that SFTs can form well beyond the primary damage region, highlighting the role of long-range vacancy and SFT diffusion [
16]. Together, these findings provide indirect validation for the mechanisms proposed in our study and affirm the broader relevance and transferability of our results.
It is important to note, however, that while ten empirical potentials were assessed for thermodynamic properties, the long-timescale MD simulations of larger SFT clusters were conducted using the Pot_Mendelev potential alone. This potential was selected due to its strong agreement with ab initio results and elasticity theory, offering superior performance in predicting formation and binding energies of SFTs. Nevertheless, the dynamic evolution of SFTs—especially for larger clusters—may still be sensitive to the choice of interatomic potential. Therefore, further studies employing improved potentials or experimental techniques such as in situ transmission electron microscopy are necessary to validate the generality of these findings.
In addition, although this study primarily focused on the thermodynamic stability and structural evolution of SFTs in aluminum, preliminary long-timescale MD simulations (tens of nanoseconds) have revealed migration events of SFTs. However, quantitative kinetic analysis—such as determining activation barriers and diffusion coefficients—requires significantly longer simulations and extensive statistical sampling (e.g., through mean squared displacement analysis across hundreds of MD runs), which is computationally intensive. To address this challenge, future work will incorporate kinetic modeling approaches, including ab initio-informed atomistic kinetic Monte Carlo and the kinetic Activation–Relaxation Technique. These methods are expected to offer deeper insights into SFT diffusion and transformation mechanisms, and the outcomes will be presented in forthcoming studies. Moreover, in future work, we also plan to extend our investigations to include the effects of impurities, alloying elements, irradiation damage, and microstructural defects. These studies will help validate the current findings under more realistic conditions and broaden their applicability to other FCC metals.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}