
First-Principles Investigation on the Adsorption and Diffusion of Oxygen at the B2(110)–O(001) Interface in Ti2AlNb Alloys





Abstract
1. Introduction
2. Computational Methods and Models
2.1. Computational Details
2.2. Structural Models
3. Results and Discussion
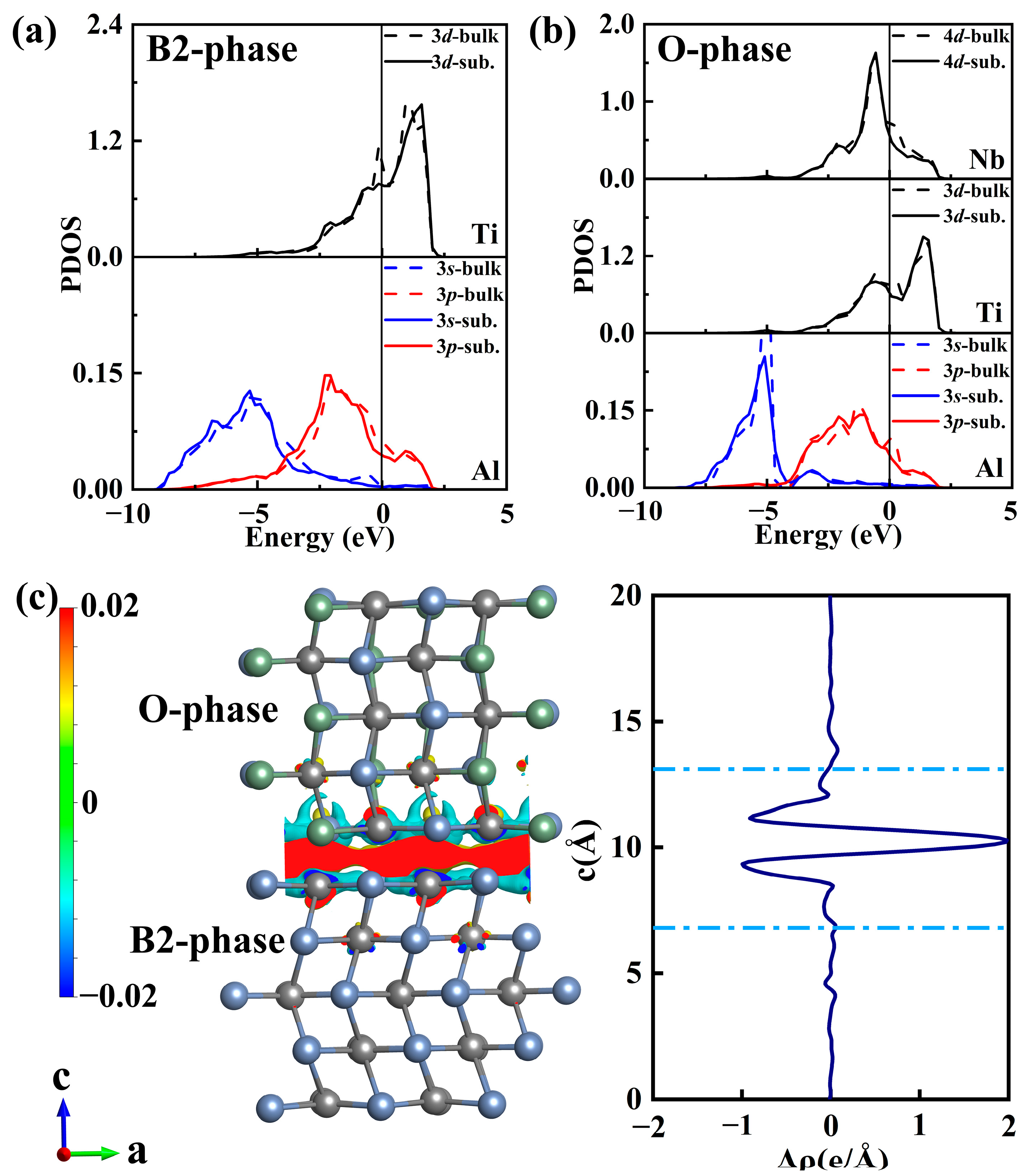
3.1. B2(110)/O(001) Interface
3.2. Oxygen Adsorption at B2(110)–O(001) Interface
3.3. Oxygen Diffusion at B2(110)–O(001) Interface
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goyal, K.; Sardana, N. Mechanical Properties of the Ti2AlNb Intermetallic: A Review. Trans. Indian Inst. Met. 2021, 74, 1839–1853. [Google Scholar] [CrossRef]
- Goyal, K.; Bera, C.; Sardana, N. Temperature-dependent structural, mechanical, and thermodynamic properties of B2-phase Ti2AlNb for aerospace applications. J. Mater. Sci. 2022, 57, 19553–19570. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Yang, Y.; Lan, X.; Zhang, Z.; Li, H. Study on Creep-Fatigue Mechanical Behavior and Life Prediction of Ti2AlNb-Based Alloy. Materials 2022, 15, 6238. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D. The intermetallic Ti2AlNb. Prog. Mater. Sci. 1997, 42, 135–158. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Yan, N.; Liang, H.Y.; Liu, Y.C. Phase transformation and microstructure control of Ti2AlNb-based alloys: A review. J. Mater. Sci. Technol. 2021, 80, 203–216. [Google Scholar] [CrossRef]
- Dai, J.; Sun, C.; Wang, A.; Zhang, H.; Li, S.; Zhang, H. High temperature oxidation and hot corrosion behaviors of Ti2AlNb alloy at 923 K and 1023 K. Corros. Sci. 2021, 184, 109336. [Google Scholar] [CrossRef]
- He, Y.S.; Hu, R.; Luo, W.Z.; He, T.; Liu, X.H. Oxidation behavior of a novel multi-element alloyed Ti2AlNb-based alloy in temperature range of 650–850 degrees C. Rare Met. 2018, 37, 838–845. [Google Scholar] [CrossRef]
- Chen, W.; Huang, L.; Liu, Y.Y.; Zhao, Y.F.; Wang, Z.; Xie, Z.W. Oxidative Corrosion Mechanism of Ti2AlNb-Based Alloys during Alternate High Temperature-Salt Spray Exposure. Coatings 2022, 12, 1374. [Google Scholar] [CrossRef]
- Qu, S.J.; Tang, S.Q.; Feng, A.H.; Feng, C.; Shen, J.; Chen, D.L. Microstructural evolution and high-temperature oxidation mechanisms of a titanium aluminide based alloy. Acta Mater. 2018, 148, 300–310. [Google Scholar] [CrossRef]
- Cheng, J.; Li, J.; Rao, Q. Time-resolved in-situ XRD study on oxidation evolution of Ti2AlNb-based alloys. Mater. Today Commun. 2023, 36, 106660. [Google Scholar] [CrossRef]
- Malecka, J. Investigation of the oxidation behavior of orthorhombic Ti2AlNb alloy. J. Mater. Eng. Perform. 2015, 24, 1834–1840. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Q.; Qu, S.J.; Xiang, H.P.; Wang, C.; Gao, J.B.; Feng, A.H.; Chen, D.L. Oxidation mechanisms of an intermetallic alloy at high temperatures. Scr. Mater. 2021, 199, 113852. [Google Scholar] [CrossRef]
- Ralison, A.; Dettenwanger, F.; Schutze, M. Oxidation of orthorhombic Ti2AlNb alloys in the temperature range 550-1000 degrees C in air. Mater. High Temp. 2003, 20, 607–629. [Google Scholar] [CrossRef]
- Xiang, J.M.; Mi, G.B.; Qu, S.J.; Huang, X.; Chen, Z.; Feng, A.H.; Shen, J.; Chen, D.L. Thermodynamic and microstructural study of Ti2AlNb oxides at 800 °C. Sci. Rep. 2018, 8, 12761. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, M.C.; Zhang, Y.R.; Liu, Y.C.; Ma, Z.Q.; Li, C.; Li, H.J. Precipitation behavior of Widmanstatten O phase associated with interface in aged Ti2AlNb-based alloys. Mater. Charact. 2018, 145, 413–422. [Google Scholar] [CrossRef]
- Zheng, Y.P.; Zeng, W.D.; Li, D.; Xu, J.W.; Ma, X.; Liang, X.B.; Zhang, J.W. Orthorhombic precipitate variant selection in a Ti2AlNb based alloy. Mater. Des. 2018, 158, 46–61. [Google Scholar] [CrossRef]
- Wang, L.; Shang, J.X.; Wang, F.H.; Chen, Y.; Zhang, Y. Oxygen adsorption on γ-TiAl surfaces and the related surface phase diagrams: A density-functional theory study. Acta Mater. 2013, 61, 1726–1738. [Google Scholar] [CrossRef]
- Wei, L.J.; Guo, J.X.; Dai, X.H.; Guan, L.; Wang, Y.L.; Liu, B.T. First-principles calculations of oxygen adsorption on the Ti3Al (0001) surface. Surf. Interface Anal. 2016, 48, 1337–1340. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkov, S.S.; Kulkova, S.E. Diffusion properties of oxygen in the α2-Ti3Al alloy. Intermetallics 2021, 137, 107281. [Google Scholar] [CrossRef]
- Li, Y.; Dai, J.H.; Song, Y.; Yang, R. Adsorption properties of oxygen atom on the surface of Ti2AlNb by first principles calculations. Comput. Mater. Sci. 2017, 139, 412–418. [Google Scholar] [CrossRef]
- Li, D.H.; Wang, B.B.; Luo, L.S.; Li, X.W.; Xu, Y.J.; Li, B.Q.; Wang, L.; Liu, W.Y.; Han, B.S.; Su, Y.Q.; et al. The interface structure and its impact on the mechanical behavior of TiAl/ Ti2AlNb laminated composites. Mater. Sci. Eng. A-Struct. Mater. Prop. Microstruct. Process. 2021, 827, 142095. [Google Scholar] [CrossRef]
- Zou, C.X.; Li, J.S.; Zhu, L.; Zhang, Y.; Yao, G.; Tang, B.; Wang, J.; Kou, H.C.; Song, H.F.; Wang, W.Y. Electronic structures and properties of TiAl/Ti2AlNb heterogeneous interfaces: A comprehensive first-principles study. Intermetallics 2021, 133, 107173. [Google Scholar] [CrossRef]
- Ishkildin, A.D.; Kistanov, A.A.; Izosimov, A.A.; Korznikova, E.A. The nitriding effect on the stability and mechanical properties of the iron titan phase: First-principles investigation. Phys. Chem. Chem. Phys. 2023, 25, 24060–24068. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, H.B.; Zhang, Y.; Lu, G.H.; Xu, H. Effects of O in a binary-phase TiAl-Ti3Al alloy: From site occupancy to interfacial energetics. J. Phys. Condens. Matter. 2011, 23, 225504. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Nelson, R.; Ertural, C.; George, J.; Deringer, V.L.; Hautier, G.; Dronskowski, R. LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis fromprojector-augmented-wave-baseddensity-functional theory. J. Comput. Chem. 2020, 41, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Q.; Liu, P.; Xie, J.P.; Ma, D.Q.; Mao, Z.P. First-principles investigation on the atomic structure, stability and electronic property of O(001)/B2(110) interface in Ti2AlNb alloys. J. Alloys Compd. 2020, 817, 152734. [Google Scholar] [CrossRef]
- Li, Y.; Dai, J.H.; Song, Y. First-principles investigation on stability and oxygen adsorption behavior of a O/B2 interface in Ti2AlNb alloys. J. Alloys Compd. 2020, 818, 152926. [Google Scholar] [CrossRef]
- Liu, P.; Han, X.; Sun, D.; Chen, Z.; Wang, Q. Adhesion, stability and electronic properties of Ti2AlN(0001)/TiAl(111) coherent interface from first-principles calculation. Intermetallics 2018, 96, 49–57. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Site (Coord.) | E(O-system) (eV) | Eads (eV) |
|---|---|---|---|
| B2-phase sub-interface | O1a (2Ti-4Al) | −548.205 | - |
| O1b (2Ti-4Al) | −548.377 | - | |
| O2a (4Ti-2Al) | −548.090 | −3.460 | |
| O2b (4Ti-2Al) | −548.380 | −3.750 | |
| O2c (4Ti-2Al) | −548.394 | −3.764 | |
| Interface | O3a (2Ti-3Al-1Nb) | −548.698 | - |
| O3b (2Ti-3Al-1Nb) | −547.340 | −2.710 | |
| O4a (4Ti-2Al) | −548.195 | −3.565 | |
| O4b (4Ti-2Al) | −548.596 | −3.966 | |
| O5a (4Ti-1Al-1Nb) | −548.379 | −3.749 | |
| O5b (4Ti-1Al-1Nb) | −548.701 | −4.071 | |
| O-phase sub-interface | O6a (2Ti-2Al-2Nb) | −548.915 | - |
| O6b (2Ti-2Al-2Nb) | −547.820 | −3.190 | |
| O7a (4Ti-1Al-1Nb) | −548.306 | −3.676 | |
| O7b (4Ti-1Al-1Nb) | −548.377 | −3.747 | |
| O8a (4Ti-2Al) | −548.747 | −4.117 | |
| O9a (4Ti-2Nb) | −548.917 | −4.287 |
| Location | Initial (Coord.) | Final (Coord.) | Energy Barrier (eV) |
|---|---|---|---|
| B2-phase sub-int. | O2a (4Ti–2Al) | O2b (4Ti–2Al) | 1.580 |
| O2b (4Ti–2Al) | O2c (4Ti–2Al) | 1.844 | |
| Interface | O3b (2Ti–3Al–1Nb) | O5a (4Ti–1Al–1Nb) | 0.199 |
| O4a (4Ti–2Al) | O4b (4Ti–2Al) | 1.698 | |
| O5a (4Ti–1Al–1Nb) | O5b (4Ti–1Al–1Nb) | 1.518 | |
| O-phase sub-int. | O6b (2Ti–2Al–2Nb) | O7a (4Ti–1Al–1Nb) | 0.568 |
| O7a (4Ti–1Al–1Nb) | O8a (4Ti–2Al) | 1.427 | |
| O8a (4Ti–2Al) | O9a (4Ti–2Nb) | 1.667 | |
| B2-phase sub-int.—Interface | O2c (4Ti–2Al) | O5b (4Ti–1Al–1Nb) | 1.632 |
| O2b (4Ti–2Al) | O4b (4Ti–2Al) | 1.825 | |
| Interface—O-phase sub-int. | O5b (4Ti–1Al–1Nb) | O9a (4Ti–2Nb) | 1.773 |
| O4b (4Ti–2Al) | O9a (4Ti–2Nb) | 1.584 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Xiang, H.; Xu, L.; Feng, A.; Qu, S.; Chen, D. First-Principles Investigation on the Adsorption and Diffusion of Oxygen at the B2(110)–O(001) Interface in Ti2AlNb Alloys. Metals 2024, 14, 316. https://doi.org/10.3390/met14030316
Zhang M, Xiang H, Xu L, Feng A, Qu S, Chen D. First-Principles Investigation on the Adsorption and Diffusion of Oxygen at the B2(110)–O(001) Interface in Ti2AlNb Alloys. Metals. 2024; 14(3):316. https://doi.org/10.3390/met14030316
Chicago/Turabian StyleZhang, Ming, Hongping Xiang, Lin Xu, Aihan Feng, Shoujiang Qu, and Daolun Chen. 2024. "First-Principles Investigation on the Adsorption and Diffusion of Oxygen at the B2(110)–O(001) Interface in Ti2AlNb Alloys" Metals 14, no. 3: 316. https://doi.org/10.3390/met14030316
APA StyleZhang, M., Xiang, H., Xu, L., Feng, A., Qu, S., & Chen, D. (2024). First-Principles Investigation on the Adsorption and Diffusion of Oxygen at the B2(110)–O(001) Interface in Ti2AlNb Alloys. Metals, 14(3), 316. https://doi.org/10.3390/met14030316