
Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days

 , and
, and
Abstract
1. Introduction
1.1. General Context
- Their definition requires a broad spectrum of multi-disciplinary knowledge, including their representativeness;
- The selected operating conditions for the measurements may differ from the mode of analyzed corrosion phenomena;
- Their implementation may be affected by future developments in electroanalytical technology, or be imperiled by locally occurring and unforeseen phenomena.
1.2. Short Review on Corrosion Assessment Studies of Various Carbon Steels in Cox-Related Environments
- The increased temperature due to radioactive disintegration (25 °C ≤ T ≤ 90 °C);
- The wide range of redox potential values over the Pourbaix diagram owing to O2, diffusing through the near field clay rock due to excavation; H2, due to release from metal corrosion; and H2S, due to the activity of sulfate-reducing bacteria.
2. Materials and Methods
2.1. Carbon Steel API 5L X65 Characteristics and Electrodes Designed for Corrosion Studies
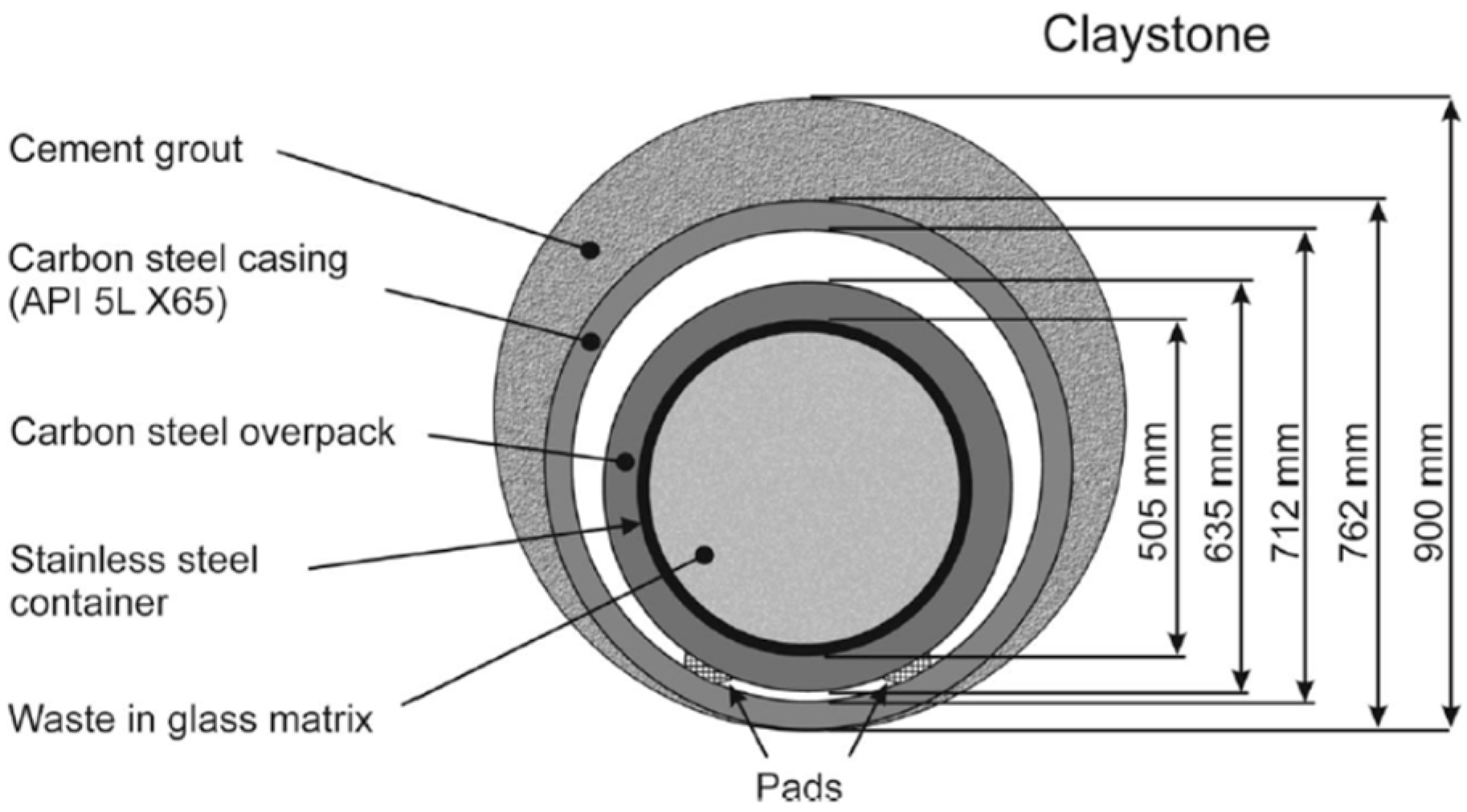
2.2. Cox Pore Water Characteristics and Borehole Device for Supplying Pore Water
2.2.1. Cox Pore Water
2.2.2. Borehole Supplying Cox Pore Water and Device for Contact with Sensors and Electrodes
2.3. Experimental Setup for Studying the Corrosivity of Cox Water against X65
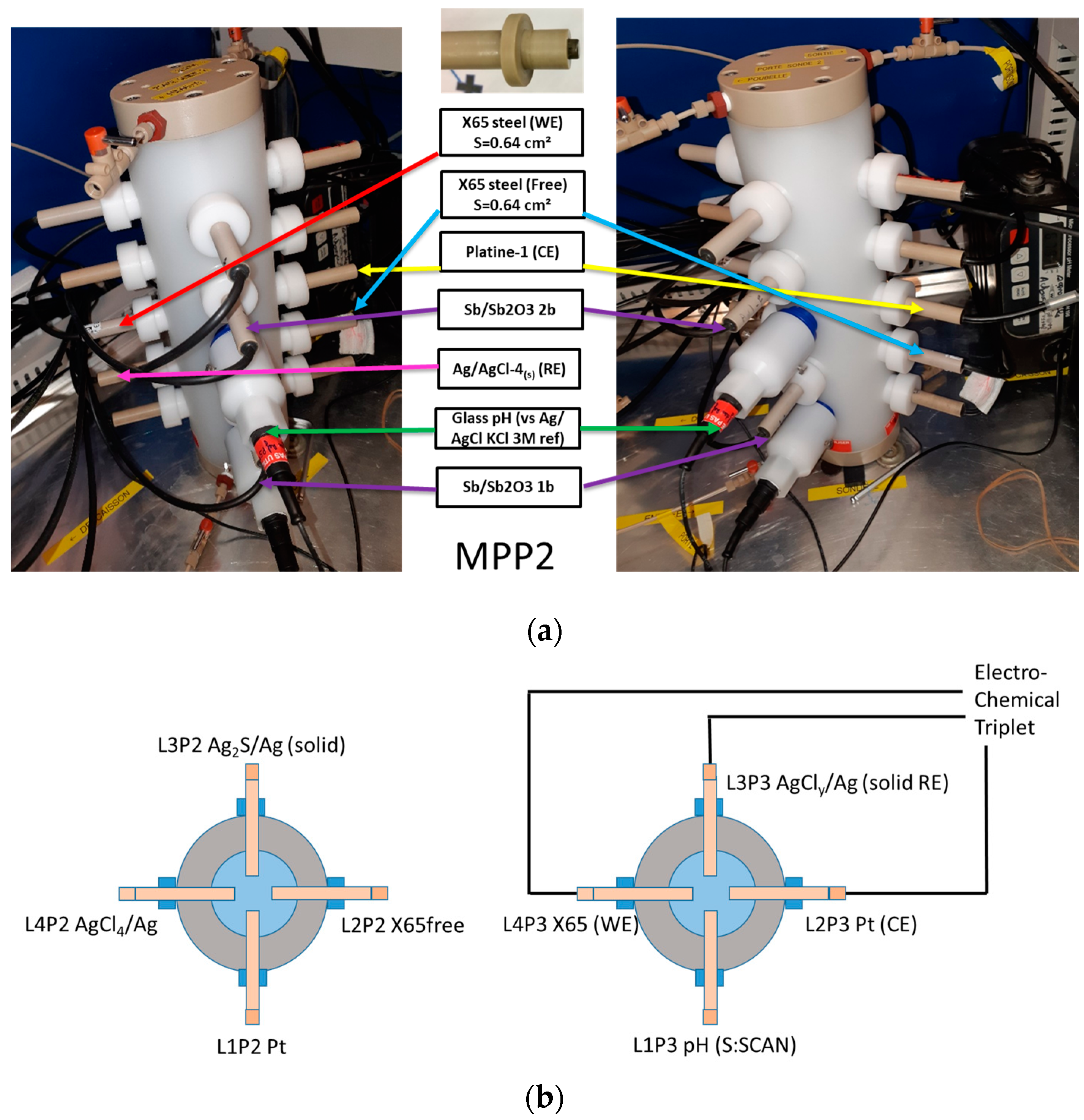
2.3.1. Multi-Parameter Probe (MPP) Holders, MPP-1 and MPP-2, and Designed Electrodes
2.3.2. Electrochemical Apparatuses, pH Meters, and Data Logger
2.3.3. Electrochemical Techniques
2.3.4. Gravimetric or Mass Loss Technique
3. Results
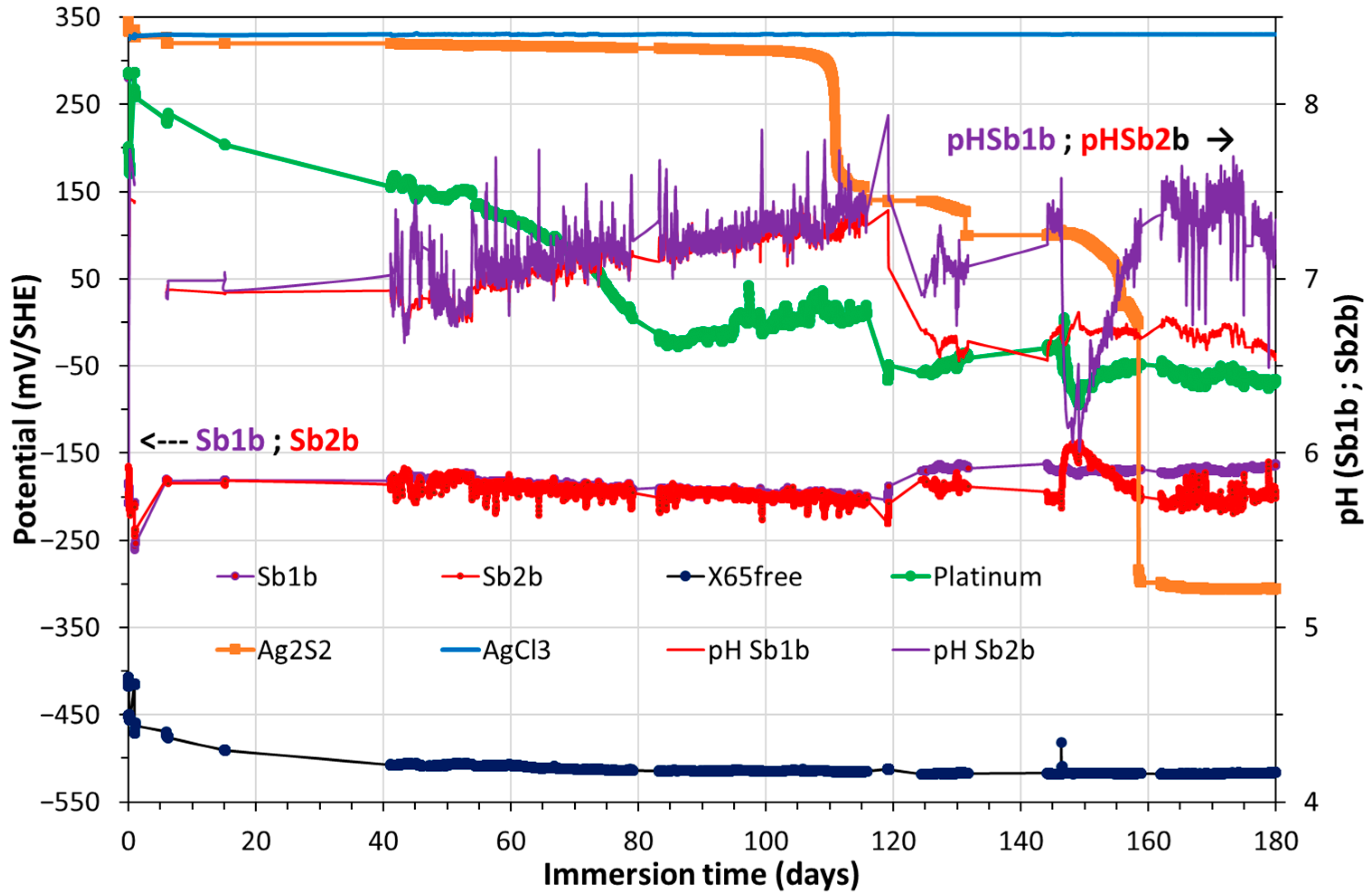
3.1. Monitoring of Temperature and OCP of Solid Reference Ag/AgCl Electrodes into MPP-2
3.2. Monitoring of OCP of Solid Electrodes, Including CS-X65 Free, into MPP-2
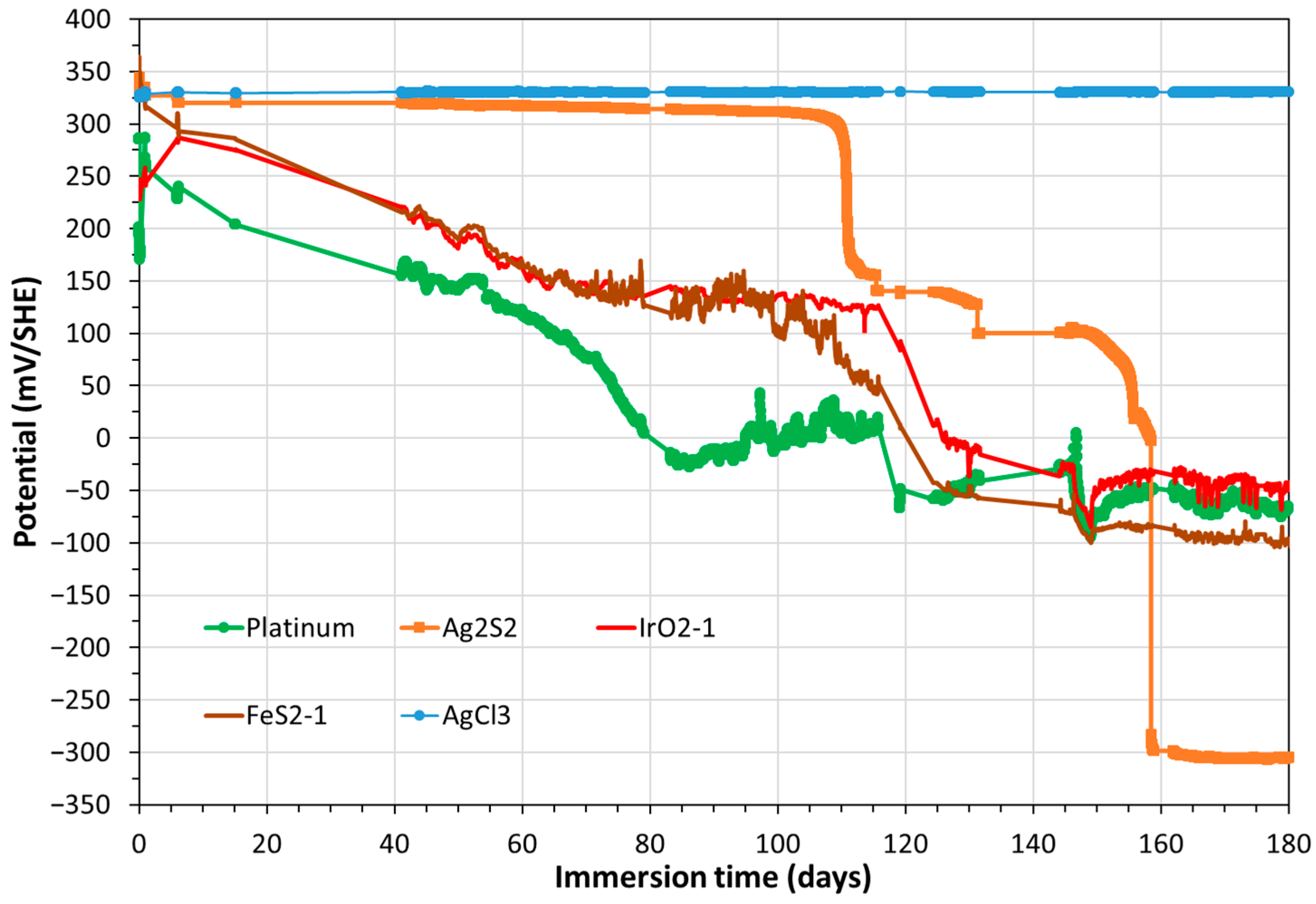
3.3. Electrochemical Corrosion Measurements on the Triplet, Including the CS-X65 Working Electrode
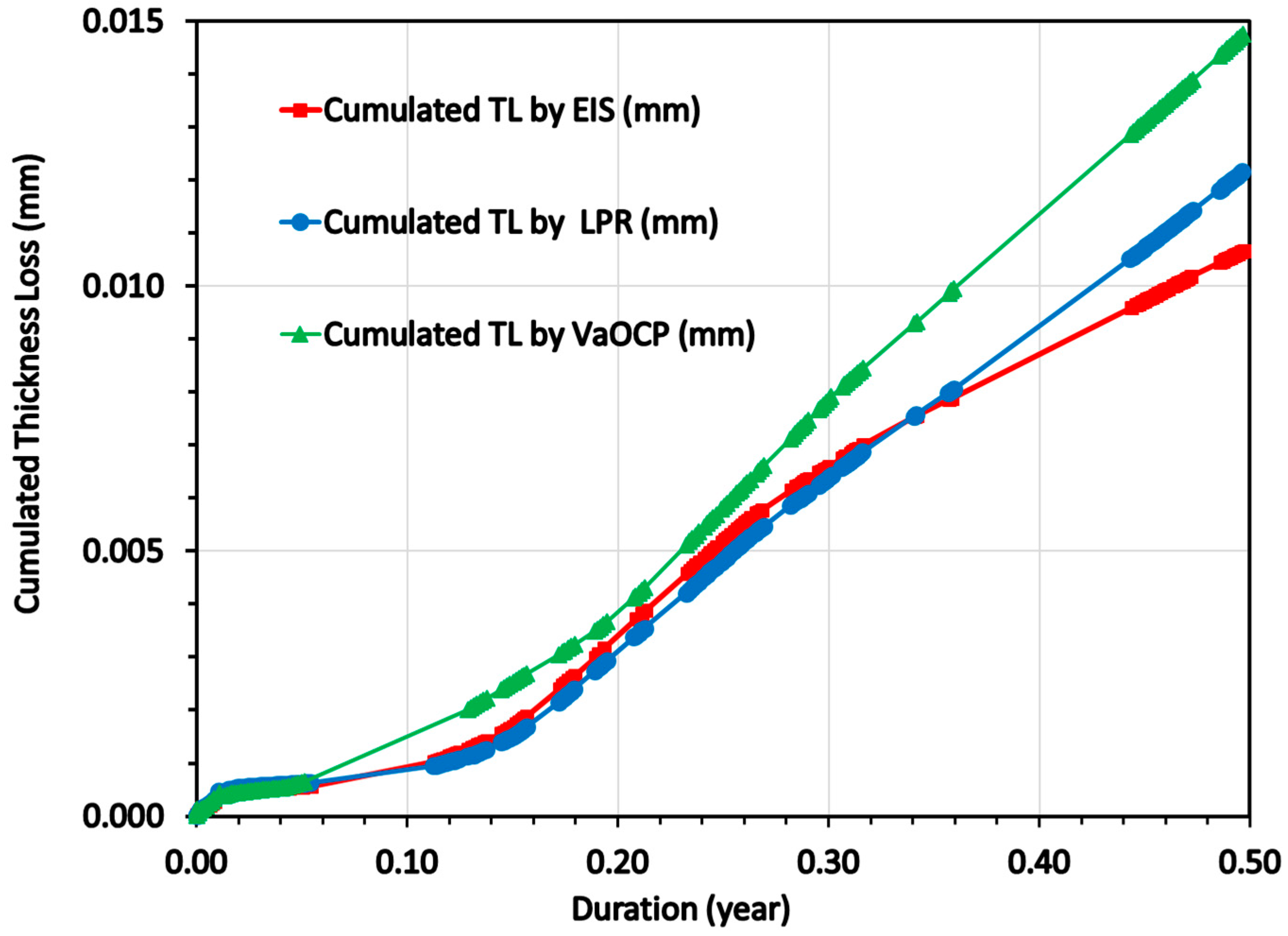
3.4. Mass Loss Corrosion Measurements on the CS-X65 Electrode (Working and Free)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B



References
- Altmann, S. “Geo”chemical research: A key building block for nuclear waste disposal safety cases. J. Contam. Hydrol. 2008, 102, 174–179. [Google Scholar] [CrossRef] [PubMed]
- French National Radioactive Waste Management Agency—Andra. Available online: https://international.andra.fr/ (accessed on 2 January 2023).
- Han, T.; Shi, J.; Chen, Y.; Li, Z. Effect of chemical corrosion on the mechanical characteristics of parent rocks for nuclear waste storage. Sci. Technol. Nucl. Install. 2016, 2016, 7853787. [Google Scholar] [CrossRef]
- Jonsson, M. Radiation effects on materials used in geological repositories for spent nuclear fuel. ISRN Mater. Sci. 2012, 2012, 639520. [Google Scholar] [CrossRef]
- Gaucher, E.C.; Robelin, C.; Matray, J.M.; Négrel, G.; Gros, Y.; Heitz, J.F.; Vinsot, A.; Rebours, H.; Cassagnabère, A.; Bouchet, A. ANDRA underground research laboratory: Interpretation of the mineralogical and geochemical data acquired in the Callovian–Oxfordian formation by investigative drilling. Phys. Chem. Earth 2004, 29, 55–77. [Google Scholar] [CrossRef]
- Gaucher, C.; Blanc, P.; Bardot, F.; Braibant, G.; Buschaert, S.; Crouzet, C.; Gautier, A.; Girard, J.-P.; Jacquot, E.; Lassin, A.; et al. Modelling the pore water chemistry of the Callovian-Oxfordian formation at a regional scale. Comptes Rendus Geosci. 2006, 338, 917–930. [Google Scholar] [CrossRef]
- Gaucher, E.; Tournassat, C.; Pearson, F.; Blanc, P.; Crouzet, C.; Lerouge, C.; Altmann, S. A robust model for pore-water chemistry of clayrock. Geochimi. Cosmochim. Acta 2009, 73, 6470–6487. [Google Scholar] [CrossRef]
- Fernández, A.M.; Sánchez-Ledesma, D.M.; Tournassat, C.; Melón, A.; Gaucher, E.C.; Astudillo, J.; Vinsot, A. Applying the squeezing technique to highly consolidated clayrocks for pore water characterization: Lessons learned from experiments at the Mont Terri Rock Laboratory. Appl. Geochem. 2014, 49, 2–21. [Google Scholar] [CrossRef]
- Tournassat, C.; Vinsot, A.; Gaucher, E.C.; Altmann, S. Chapter 3—Chemical conditions in clay-rocks Natural and Engineered Clay Barriers. In Natural and Engineered Clay Barriers, 1st ed.; Tournassat, C., Steefel, C.I., Bourg, I.C., Bergaya, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 71–100. [Google Scholar]
- Tournassat, C.; Steefel, C.I.; Bourg, I.C.; Bergaya, F. Chapter 1—Surface Properties of Clay Minerals. In Natural and Engineered Clay Barriers, 1st ed.; Tournassat, C., Steefel, C.I., Bourg, I.C., Bergaya, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 5–31. [Google Scholar]
- Vinsot, A.; Appelo, C.; Cailteau, C.; Wechner, S.; Pironon, J.; De Donato, P.; De Cannière, P.; Mettler, S.; Wersin, P.; Gäbler, H.-E. CO2 data on gas and pore water sampled in situ in the Opalinus Clay at the Mont Terri rock laboratory. Phys. Chem. Earth Parts A B C 2008, 33, 54–60. [Google Scholar] [CrossRef]
- Vinsot, A.; Mettler, S.; Wechner, S. In situ characterization of the Callovo-Oxfordian pore water composition. Phys. Chem. Earth Parts A B C 2008, 33, S75–S86. [Google Scholar] [CrossRef]
- Betelu, S.; Ignatiadis, I.; Tournassat, C. Redox potential measurements in a claystone. In Proceedings of the Clay Conference, Davos, Switzerland, 24–27 September 2017; p. 111. [Google Scholar]
- Betelu, S.; Ignatiadis, I.; Tournassat, C. Redox potential measurements in Callovian Oxfordian claystone: Insights into the nature of the mineral(s) responsible for the redox reactivity. In Proceedings of the Clay Conference, Nancy, France, 13–16 June 2022; p. Clay00387. [Google Scholar]
- Abramova, E.; Popova, N.; Artemiev, G.; Boldyrev, K.; Kazakov, K.; Kryuchkov, D.; Safonov, A. Biological factors affecting the evolution of safety barrier materials in the Yeniseisky deep geological repository. Eng. Geol. 2023, 312, 106931. [Google Scholar] [CrossRef]
- Fernández, A.M.; Kaufhold, S.; Olin, M.; Zheng, L.G.; Wersin, P.; Wilson, J. Editorial for Special Issue “Clay Mineral Transformations after Bentonite/Clayrocks and Heater/Water Interactions from Lab and Large-Scale Tests”. Minerals 2022, 12, 569. [Google Scholar] [CrossRef]
- Karthick, S.; Muralidharan, S.; Saraswathy, V. Corrosion performance of mild steel and galvanized iron in clay soil environment. Arab. J. Chem. 2020, 13, 3301–3318. [Google Scholar] [CrossRef]
- Schlegel, M.L.; Martin, F.; Fenart, M.; Blanc, C.; Varlet, J.; Foy, E. Corrosion of carbon steel in clay compact environments at 90 °C: Effect of confined conditions. Corros. Sci. 2021, 184, 109368. [Google Scholar] [CrossRef]
- Leupin, O.X.; Smart, N.R.; Zhang, Z.; Stefanoni, M.; Angst, U.; Papafotiou, A.; Diomidis, N. Anaerobic corrosion of carbon steel in bentonite: An evolving interface. Corros. Sci. 2021, 187, 109523. [Google Scholar] [CrossRef]
- Chaparro, M.C.; Finck, N.; Metz, V.; Geckeis, H. Reactive Transport Modelling of the Long-Term Interaction between Carbon Steel and MX-80 Bentonite at 25 °C. Minerals 2021, 11, 1272. [Google Scholar] [CrossRef]
- Shrestha, R.; Černoušek, T.; Stoulil, J.; Kovářová, H.; Sihelská, K.; Špánek, R.; Ševců, A.; Steinová, J. Anaerobic microbial corrosion of carbon steel under conditions relevant for deep geological repository of nuclear waste. Sci. Total Environ. 2021, 800, 149539. [Google Scholar] [CrossRef]
- Reddy, B.; Padovani, C.; Smart, N.R.; Rance, A.P.; Cook, A.; Milodowski, A.; Field, L.; Kemp, S.; Diomidis, N. Further results on the in situ anaerobic corrosion of carbon steel and copper in compacted bentonite exposed to natural Opalinus Clay porewater containing native microbial populations. Mater. Corros. 2021, 72, 268–281. [Google Scholar] [CrossRef]
- Crusset, D.; Deydier, V.; Necib, S.; Gras, J.M.; Combrade, P.; Féron, D.; Burger, E. Corrosion of carbon steel components in the French high-level waste programme: Evolution of disposal concept and selection of materials. Corros. Eng. Sci. Technol. 2017, 52, 17–24. [Google Scholar] [CrossRef]
- Martin, F.A.; Perrin, S.; Bataillon, C. Evaluating the corrosion rate of low alloyed steel in Callovo-Oxfordian clay: Towards a complementary EIS, gravimetric and structural study. MRS Online Proc. Libr. 2012, 1475, 471–476. [Google Scholar] [CrossRef]
- Schlegel, M.L.; Necib, S.; Daumas, S.; Blanc, C.; Foy, E.; Trcera, N.; Romaine, A. Microstructural characterization of carbon steel corrosion in clay borehole water under anoxic and transient acidic conditions. Corros. Sci. 2016, 109, 126–144. [Google Scholar] [CrossRef]
- Necib, S.; Linard, Y.; Crusset, D.; Schlegel, M.L.; Daumas, S.; Michau, N. Corrosion processes of C-steel in long-term repository conditions. Corros. Eng. Sci. Technol. 2017, 52, 127–130. [Google Scholar] [CrossRef]
- Necib, S.; Linard, Y.; Crusset, D.; Michau, N.; Daumas, S.; Burger, E.; Romaine, A.; Schlegel, M.L. Corrosion at the carbon steel clay borehole water and gas interfaces at 85 °C under anoxic and transient acidic conditions. Corros. Sci. 2016, 111, 242–258. [Google Scholar] [CrossRef]
- El Hajj, H.; Abdelouas, A.; Grambow, B.; Martin, C.; Dion, M. Microbial corrosion of P235GH steel under geological conditions. Phys. Chem. Earth 2010, 35, 248–253. [Google Scholar] [CrossRef]
- El Mendili, C.Y.; Abdelouas, A.; Bardeau, J.F. Corrosion of P235GH carbon steel in simulated Bure soil solution. J. Mater. Environ. Sci. 2013, 4, 786–791. [Google Scholar]
- El Mendili, Y.; Abdelouas, A.; Karakurt, G.; Chaou, A.A.; Essehli, R.; Bardeau, J.-F.; Greneche, J.-M. The effect of temperature on carbon steel corrosion under geological conditions. Appl. Geochem. 2015, 52, 76–85. [Google Scholar] [CrossRef]
- Daoudi, J.; Betelu, S.; Tzedakis, T.; Bertrand, J.; Ignatiadis, I. A Multi-Parametric Device with Innovative Solid Electrodes for Long-Term Monitoring of pH, Redox-Potential and Conductivity in a Nuclear Waste Repository. Sensors 2017, 17, 1372. [Google Scholar] [CrossRef]
- Lundy, M.; Garitte, B.; Lettry, Y.; Vinsot, A. Water Rock Interaction [WRI 14] Experimental design for in situ characterization of the Callovo-Oxfordian pore water composition at 85 °C. Procedia Earth Planet. Sci. 2013, 7, 533–536. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals & Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2000; ISBN 978-0-471-04372-0. [Google Scholar]
- Rodrigues, R.; Gaboreau, S.; Gance, J.; Ignatiadis, I.; Betelu, S. Reinforced concrete structures: A review of corrosion mechanisms and advances in electrical methods for corrosion monitoring. Constr. Build. Mater. 2021, 269, 121240. [Google Scholar] [CrossRef]
- Fahim, A.; Ghods, P.; Isgor, O.B.; Thomas, M.D.A. A critical examination of corrosion rate measurement techniques applied to reinforcing steel in concrete. Mater. Corros. 2018, 69, 1784–1799. [Google Scholar] [CrossRef]
- Ribeiro, D.V.; Abrantes, J.C.C. Application of electrochemical impedance spectroscopy (EIS) to monitor the corrosion of reinforced concrete: A new approach. Constr. Build. Mater. 2016, 111, 98–104. [Google Scholar] [CrossRef]
- Stern, M.; Geary, A.L. Electrochemical polarization: I. A theoretical analysis of the shape of polarization curves. J. Electrochem. Soc. 1957, 104, 56–63. [Google Scholar] [CrossRef]
- Andrade, C.; Alonso, C. Test methods for on-site corrosion rate measurement of steel reinforcement in concrete by means of the polarization resistance method. Mater. Struct. 2004, 37, 623–643. [Google Scholar] [CrossRef]
- Ignatiadis, I.; Amalhay, M. Experimental study of corrosion of two stainless steels during the oxidative bacterial dissolution of pyrite using mass loss and electrochemical techniques. Int. J. Miner. Process. 2001, 62, 199–215. [Google Scholar] [CrossRef]
- ASTM G16-13(2019); Standard Guide for Applying Statistics to Analysis of Corrosion Data. ASTM International: West Conshohocken, PA, USA, 2019.
- Haynie, F.H. Statistical treatment of data, data interpretation and reliability. In Corrosion Tests and Standards: Application and Interpretation, 2nd ed.; Baboian, R., Ed.; ASTM International: West Conshohocken, PA, USA, 2005; pp. 83–88. [Google Scholar]
- Roberge, P.R. Statistical interpretation of corrosion test results. In ASM Handbook: Corrosion: Fundamentals, Testing and Protection, 10th ed.; Cramer, S.D., Covino, B.S., Eds.; ASM International: West Conshohocken, PA, USA, 2003; pp. 425–429. [Google Scholar]
- Panchenko, Y.M.; Marshakov, A.I.; Igonin, T.N.; Kovtanyuk, V.V.; Nikolaeva, L.A. Long-term forecast of corrosion mass losses of technically important metals in various world regions using a power function. Corros. Sci. 2014, 88, 306–316. [Google Scholar] [CrossRef]
- Amalhay, M.; Abou Akar, A.; Ignatiadis, I. Overview of the deposition phenomena in geothermal wells in the Paris Basin. In Proceedings of the World Geothermal Congress, Worldwide Utilization of Geothermal Energy; Barbier, E., Frye, G., Iglesias, E., Palmason, G., Eds.; International Geothermal Association (IGA): Auckland, New Zealand, 1994; pp. 2497–2502. [Google Scholar]
- Amalhay, M.; Cotiche, C.; Ignatiadis, I. Study of the electrochemical behavior of a carbon-steel in a real and treated geothermal environment. In Proceedings of the World Geothermal Congress, Worldwide Utilization of Geothermal Energy; Barbier, E., Frye, G., Iglesias, E., Palmason, G., Eds.; International Geothermal Association (IGA): Auckland, New Zealand, 1995; Volume 4, pp. 2369–2374. [Google Scholar]
- Amalhay, M.; Ignatiadis, I. Study of the behavior of a carbon-steel in a natural or modified and treated geothermal environment. In Proceedings of the International Symposium, Eurocorr’ 96, Nice, France, 24–26 September 1996; pp. 1–4. [Google Scholar]
- Ignatiadis, I.; Amalhay, M. The effect of iron sulfide deposits on corrosion of carbon steel in a natural and treated geothermal water using electrochemical techniques and X-ray diffractometry. In Proceedings of the International Symposium, Eurocorr’ 96, Nice, France, 24–26 September 1996; pp. 1–4. [Google Scholar]
- Rickard, D.; Luther, G.W. Chemistry of Iron Sulfides. Chem. Rev. 2007, 107, 514–562. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | Chemical Composition (% Mass) |
|---|---|
| C | 0.16 |
| Mn | 1.65 |
| Si | 0.45 |
| Ti | 0.06 |
| P | 0.02 |
| S | 0.01 |
| V | 0.07 |
| Nb | 0.05 |
| Fe | ~97.53 |
| Elements or Chemistry | Concentration (mM) or Value | Elements | Concentration (mM) |
|---|---|---|---|
| pH | 7.35 | K | 7.07 |
| Redox potential (mV/SHE) | −180 | Ca | 14.8 |
| Ionic strength | 116.0 | Mg | 14.1 |
| C(4) | 1.29 | Sr | 1.12 |
| S− | 34.0 | Si | 0.0943 |
| Cl− | 30.1 | Fe | 0.0940 |
| Na | 32.0 | Al | 0.0000086 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano Moyeme, Y.C.; Betelu, S.; Bertrand, J.; Groenen Serrano, K.; Ignatiadis, I. Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days. Metals 2023, 13, 966. https://doi.org/10.3390/met13050966
Sano Moyeme YC, Betelu S, Bertrand J, Groenen Serrano K, Ignatiadis I. Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days. Metals. 2023; 13(5):966. https://doi.org/10.3390/met13050966
Chicago/Turabian StyleSano Moyeme, Yendoube Charles, Stephanie Betelu, Johan Bertrand, Karine Groenen Serrano, and Ioannis Ignatiadis. 2023. "Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days" Metals 13, no. 5: 966. https://doi.org/10.3390/met13050966
APA StyleSano Moyeme, Y. C., Betelu, S., Bertrand, J., Groenen Serrano, K., & Ignatiadis, I. (2023). Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days. Metals, 13(5), 966. https://doi.org/10.3390/met13050966