
Effect of Segregation of Sc, Y and La Atoms on Prenucleation at the Liquid-Al/γ-Al2O3{1 1 1} Interfaces



Abstract
1. Introduction
2. Methods
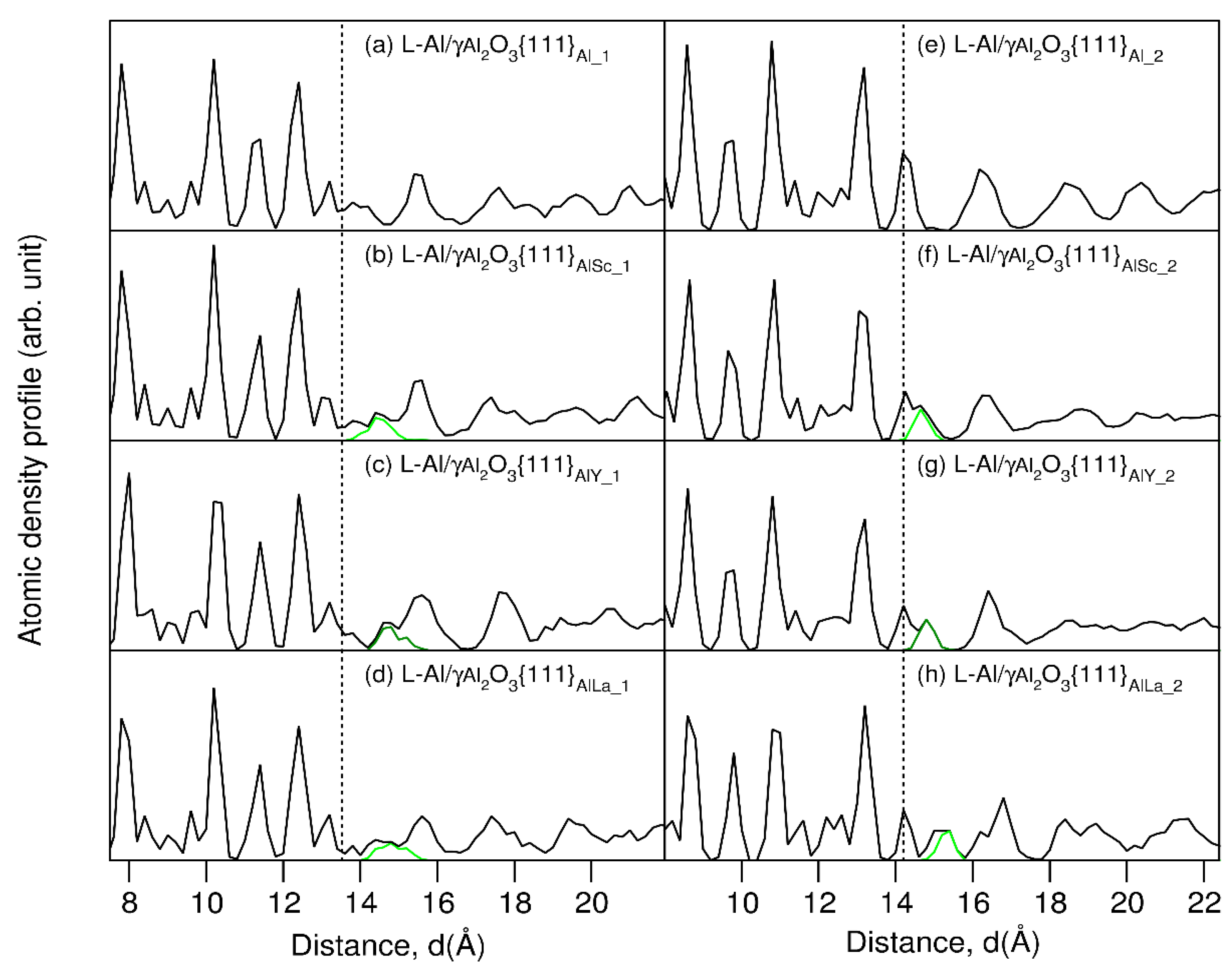
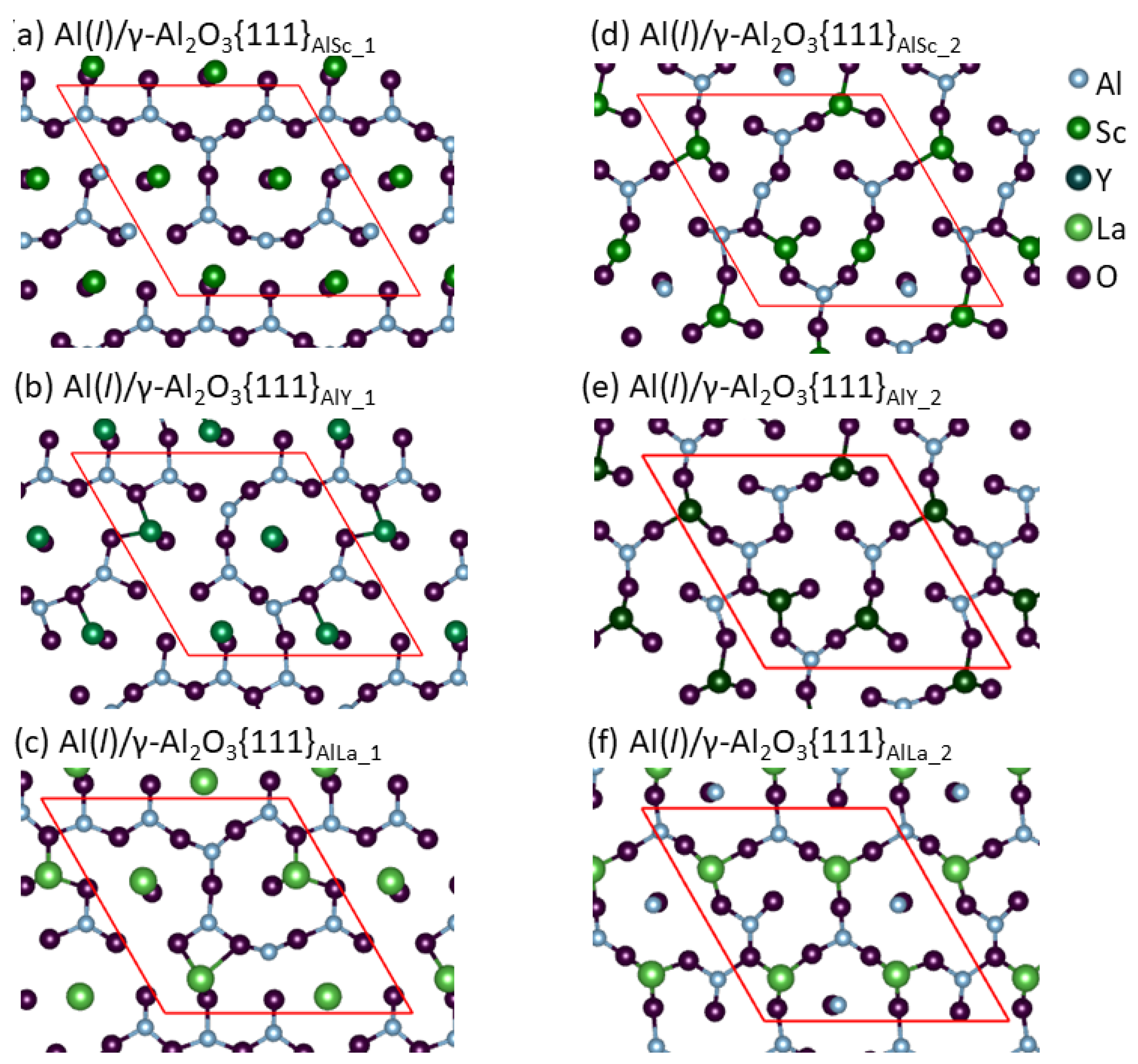
3. Results
4. Discussion
5. Conclusions
- (1)
- The Group 3 element segregated Al(l)/γ-Al2O3{1 1 1} systems exhibit high stability, corresponding to the higher formation energies of the Group 3 metal oxides than that of alumina.
- (2)
- Segregation of the Group 3 atoms causes reconstructions of the metal layers adjacent to the γ-Al2O3{1 1 1} substrates and atomic roughness of the substrate surfaces. The widths of the metal layers are proportional to the ionic radii of the segregated Group 3 ions.
- (3)
- The segregated Group 3 atoms/ions exhibit strong localization, which corresponds to their strong bonding to the outmost O.
- (4)
- The atomic ordering (prenucleation) at the Group 3 atoms segregated Al(l)/γ-Al2O3{1 1 1} interfaces is reduced to the same level. This maximizes the number of potential nucleation sites during solidification.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Impey, S.A.; Stephenson, D.J.; Nicholls, J.R. Mechanism of scale growth on liquid aluminium. J. Mater. Sci. Technol. 1988, 4, 1126–1132. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.-T.; Fan, Z. Oxidation of aluminium alloy melt and inoculation by oxide particles. Trans. Indian Inst. Met. 2012, 65, 653–661. [Google Scholar] [CrossRef]
- Li, H.-T.; Wang, Y.; Fan, Z. Mechanisms of enhanced heterogeneous nucleation during solidification in binary Al-Mg alloys. Acta Mater. 2012, 60, 1528–1537. [Google Scholar] [CrossRef]
- Kim, K. Formation of endogenous MgO and MgAl2O4 particles and their possibility of acting as substrate for heterogeneous nucleation of aluminum grains. Surf. Interface Anal. 2015, 47, 429–438. [Google Scholar] [CrossRef]
- Fang, C.M.; Fan, Z. Ab Initio MD Investigation of Prenucleation at Liquid-Metal/Oxide Interfaces: An Overview. Metals 2022, in progress. [Google Scholar]
- Verwey, E.J.W. The crystal structure of γ-Fe2O3 and γ-Al2O3. Z. Kristallogr. 1935, 91, 65–69. [Google Scholar] [CrossRef]
- Smrčok, L.; Langer, L.; Krestan, J. γ-Alumina: A single crystal X-ran diffraction study. Acta Crystallogr. 2006, C62, i83–i84. [Google Scholar] [CrossRef]
- Fang, C.M.; Yasmin, S.; Fan, Z. Interfacial interaction and prenucleation at liquid-Al/γ-Al2O3{1 1 1} interfaces from ab initio molecular dynamics simulations. J. Phys. Commun. 2021, 5, 015007. [Google Scholar] [CrossRef]
- Men, H.; Fan, Z. Prenucleation induced by crystalline substrates. Metall. Mater. Trans. A 2018, 49, 2766–2777. [Google Scholar] [CrossRef]
- Fang, C.M.; Men, H.; Fan, Z. Effect of substrate chemistry on prenucleation. Metall. Mater. Trans. A 2018, 49, 4242–6231. [Google Scholar] [CrossRef]
- Men, H.; Fang, C.M.; Fan, Z. Prenucleation at the liquid/substrate interface: An overview. Metals 2022, in progress. [Google Scholar]
- Fan, Z., Mendis, C., Eds.; Heterogeneous nucleation, grain initiation and grain refinement of Mg-alloys. In Proceedings of the 11th International Conference on Magnesium Alloys and Their Applications, Beaumont Estate, Old Windsor, UK, 24–27 July 2018; p. 7. [Google Scholar]
- Fan, Z.; Gao, F.; Jiang, B.; Que, Z.P. Impeding nucleation for more significant grain refinement. Sci. Rep. 2020, 10, 9448. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Gao, F. Grain initiation and grain refinement: An overview. Metals 2022, in progress. [Google Scholar]
- Kelton, K.F.; Greer, A.L. Nucleation in Condensed Matter: Applications in Materials and Biology; Pergamon Materials Series; Elsevier Ltd.: Oxford, UK; Amsterdam, The Netherlands, 2010. [Google Scholar]
- Cibula, A. The grain refinement of aluminium alloy castings by addition of titanium and boron. J. Inst. Met. 1951, 80, 1–16. [Google Scholar]
- Greer, A.L.; Bunn, A.M.; Tronche, A.; Evans, P.V.; Bristo, D.J. Modelling of inoculation of metallic melts: Application to grain refinement of aluminium by Al-Ti-B. Acta Mater. 2000, 48, 2823–2835. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Zhang, Y.; Qin, T.; Zhou, X.R.; Thompson, G.E.; Pennycook, T.; Hashimoto, T. Grain refining mechanism in the Al/Al-Ti-B system. Acta Mater. 2015, 84, 292–304. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, C.M.; Zhou, L.; Hashimoto, T.; Zhou, X.R.; Ramasse, Q.M.; Fan, Z. Zr poisoning of TiB2 grain refinement. Acta Mater. 2019, 164, 428–439. [Google Scholar] [CrossRef]
- Lide, D.R. (Editor-in-Chief) CRC Handbook of Chemistry and Physics, 84th ed.; CRC Press: Cleveland, OH, USA, 2003; pp. 4–112. [Google Scholar]
- Wang, Y.; Wang, S.H.; Fang, C.M.; Que, Z.P.; Hashimoto, T.; Zhou, X.R.; Ramasse, Q.M.; Fan, Z. Manipulating nucleation potency of substrates by interfacial segregation: An overview. Metals 2022, in progress. [Google Scholar]
- Wang, Y.; Que, Z.P.; Hashimoto, T.; Zhou, X.R.; Fan, Z. Mechanism for Si poisoning of Al-Ti-B grain refiners in Al alloys. Metall. Mater. Trans. A 2020, 51, 5734–5757. [Google Scholar] [CrossRef]
- Easton, M.A.; Qian, M.; Prasad, A.; StJohn, D.H. Recent advances in grain refinement of light metals and alloys. Curr. Opin. Solid State Mater. Sci. 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Liu, Z. Review of grain refinement of cast metals through inoculation: Theories and developments. Metall. Mater. Trans. A 2017, 48, 4755–4776. [Google Scholar] [CrossRef]
- Fiquet, G.; Richet, P.; Montagnac, G. High-temperature thermal expansion of lime, periclase, corundum and spinel. Phys. Chem. Min. 1999, 27, 103–111. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17978. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Brostow, W.; Hagg Lodbland, H.E. Materials: Introduction and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Fang, C.M.; Mohammadi, V.; Nihtianov, S.; Sluiter, M.F.H. Stability, geometry and electronic properties of BHn (n = 0 to 3) radicals on the Si(001)3 × 1:H surface from first-principles. J. Phys. Cond. Matter 2020, 32, 235201. [Google Scholar] [CrossRef]
- Arblaster, J.W. Selected Values of the Crystallographic Properties of the Elements; ASM International: Materials Park, OH, USA, 2018; pp. 118–228. [Google Scholar]
- Hashibon, A.; Adler, J.; Finnis, M.W.; Kaplan, W.D. Atomistic study of structural correlations at a liquid-solid interface. Comp. Mater. Sci. 2002, 24, 443–452. [Google Scholar] [CrossRef]
- Fan, Z. An epitaxial model for heterogeneous nucleation on potent substrate. Metall. Mater. Trans. A 2013, 44, 1409–1418. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum-theory of molecular-structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. A bonded path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. The Structure of Crystals, 2nd ed.; Reinhold Publishing Corporation: New York, NY, USA, 1935. [Google Scholar]
- Knop, O.; Hartley, J.M. Refinement of the crystal structure of scandium oxide. Can. J. Chem. 1968, 46, 1446–1450. [Google Scholar] [CrossRef]
- Hanic, F.; Hartmanová, M.; Knab, G.G.; Urusovskaya, A.A.; Bagdasarov, K.S. Real structure of undoped Y2O3 single crystals. Acta Crystallogr. B 1984, 40, 76–82. [Google Scholar] [CrossRef]
- Sutton, L.E. (Ed.) Tables of Interatomic Distances and Configuration in Molecules and Ions; The Chemical Society: London, UK, 1958. [Google Scholar]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–922. [Google Scholar] [CrossRef]
- Ellingham, H.J.T. Reducibility of oxides and sulphides in metallurgical processes. J. Soc. Chem. Ind. Trans. Commun. 1944, 63, 125–160. [Google Scholar]
- Rafigue, M.M.A. Pyrometallurgy and electrometallurgy of rare earths—Part A: Analysis of metallothermic reduction and its variants. Preprints 2021, 2021030463. [Google Scholar] [CrossRef]
- Van Der Geest, A.G.; Kolmogorov, A.N. Stability of 41 metal-boron systems at 0 GPa and 30 GPa from first principles. Calphad 2014, 46, 184–204. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Ma, S.D.; Dong, Z.H.; Zong, N.F.; Jing, T.; Dong, H.B. Solute-adsorption enhanced heterogeneous nucleation: The effect of Cu adsorption on α-Al at the sapphire substrate. Phys. Chem. Chem. Phys. 2021, 23, 5270–5282. [Google Scholar] [CrossRef]
- Bojarski, S.A.; Stuer, M.; Zhao, Z.; Bowen, P.; Rohere, G.S. Influence of Y and La additions on grain growth and the grain-boundary character distribution of alumina. J. Am. Ceram. Soc. 2014, 97, 622–630. [Google Scholar] [CrossRef]
- Karakozov, E.S.; Konyshov, G.V.; Musin, R.S. Fundamentals of welding metals to ceramic materials. J. Weld. Intern. 1993, 7, 991–996. [Google Scholar] [CrossRef]
- Oh, S.H.; Kauffmann, Y.; Scheu, C.; Kaplan, W.D.; Rühle, N. Ordered liquid aluminium at the sapphire. Science 2005, 310, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.D.; Chatain, D.; Wynblatt, P.; Cater, W.C. A review of wetting versus adsorption, complexions, and related phenomena: The rosetta stone of wetting. J. Mater. Sci. 2013, 48, 5681–5717. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface | f (%) (2) | Rn (%) (3) | n (Al Layers) | |||
|---|---|---|---|---|---|---|
| Al(l)/γ-Al2O3{1 1 1}AlM−1 | 4.8 | Lterm(Al) | L(M) | L1(Al) | Sum (4) | |
| Pure Al (1) | 54.0 | - | 72.2 | 126.2 | 3 | |
| (distance to O) (Å) (5) | (1.01) | - | (3.42) | - | ||
| M = Sc | 48.8 | 25 | 76.4 | 150.2 | 2 | |
| (distance to O) (Å) (5) | (0.76) | (2.04) | (3.16) | - | ||
| M = Y | 46.3 | 25 | 72.3 | 143.6 | 1~2 | |
| (distance to O) (Å) (5) | (0.78) | (2.28) | (3.22) | - | ||
| M = La | 40.6 | 25 | 52.5 | 118.1 | 2~3 | |
| (distance to O) (Å) (5) | (0.79) | (2.24) | (3.34) | - | ||
| Al(l)/γ-Al2O3{1 1 1}AlM−2 | 4.8 | Lterm(Al) | L(M) | L1(Al) | Sum | 0 |
| Pure Al (1) | 58.1 | - | 71.9 | 130.0 | 3~4 | |
| (distance to O) (Å) (5) | (0.83) | - | (3.43) | - | 0 | |
| M = Sc | 42.8 | 25 | 76.5 | 144.3 | 2 | |
| (distance to O) (Å) (5) | (1.12) | (1.50) | (3.27) | - | 0 | |
| M = Y | 43.3 | 25 | 72.3 | 140.6 | 1~2 | |
| (distance to O) (Å) (5) | (0.98) | (1.59) | (3.33) | - | 0 | |
| M = La | 43.8 | 25 | 54.3 | 123.1 | 2~3 | |
| (distance to O) (Å) (5) | (1.02) | (2.08) | (3.62) | - | 0 | |
| Phase | Lattice, Space Group | lattice Paras. (Å) | M-O Bonds (Å) Ionic Radius (Å) | Eform (eV/f.u.) ΔE (eV/f.u.) |
|---|---|---|---|---|
| α-Al2O3 | Rhom. R-3c(167) | a = 4.807 (4.785) [25] c = 13.116 (12.991) | Al-O: 1.86 (×3), 1.97 (×3) Aver. 1.92 0.52, (0.675) [45] | −15.10 0.0 eV |
| c-Sc2O3 | Cub. Ia-3(206) | a = 9.854 (9.8459) [38] | Sc1-O: 2.09 (×2), 2.12 (×2), 2.16 (×2) Sc2-O: 2.12 (×6) Aver. 2.12 0.72 (0.885) [45] | −17.84 −2.74 |
| c-Y2O3 | Cub. Ia-3(206) | a = 10.654 (10.600) [39] | Y1-O: 2.26 (×2), 2.28 (×2), 2.35 (×2) Y2-O: 2.29 (×6) Aver. 2.30 0.90 (1.040) [45] | −18.09 −2.99 |
| c-La2O3 | Cubic. Ia-3(206) | a = 11.397 (11.360) [37] | La1-O: 2.42 (×2), 2.43 (×2), 2.50 (×2) La2-O: 2.45 (×6) Aver. 2.45 1.05 (1.172) [45] | −17.36 −2.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, C.; Fan, Z. Effect of Segregation of Sc, Y and La Atoms on Prenucleation at the Liquid-Al/γ-Al2O3{1 1 1} Interfaces. Metals 2022, 12, 1550. https://doi.org/10.3390/met12101550
Fang C, Fan Z. Effect of Segregation of Sc, Y and La Atoms on Prenucleation at the Liquid-Al/γ-Al2O3{1 1 1} Interfaces. Metals. 2022; 12(10):1550. https://doi.org/10.3390/met12101550
Chicago/Turabian StyleFang, Changming, and Zhongyun Fan. 2022. "Effect of Segregation of Sc, Y and La Atoms on Prenucleation at the Liquid-Al/γ-Al2O3{1 1 1} Interfaces" Metals 12, no. 10: 1550. https://doi.org/10.3390/met12101550
APA StyleFang, C., & Fan, Z. (2022). Effect of Segregation of Sc, Y and La Atoms on Prenucleation at the Liquid-Al/γ-Al2O3{1 1 1} Interfaces. Metals, 12(10), 1550. https://doi.org/10.3390/met12101550