
First-Principles Study on the Mechanism of Greenhouse Gas Generation in Aluminum Electrolysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. First-Principles Calculation
2.1.1. Computational Hypothesis
- The total energy change of O or F of the Al-F complex ion [F]− and Al-O-F complex ion [O]2− in molten salt moving from the electrolyte to the anode surface for discharge, transferring charge and interacting with the anode is: ∆E = ∆Eef + ∆Eet + ∆Ebf, where ∆Eef is the work of the electric field to [O]2− and [F]−, ∆Eet is the energy change caused by the electron transfer of the electric field to [O]2− and [F]− on the anode surface, and ∆Ebf is the energy change of Oanode and Fanode formed by the interaction of O and F atoms with the anode;
- The surface potential of the anode on an atomic scale is equal. When [O]2− and [F]− with the same number of charges move from the inside of the electrolyte to any position of the anode, the electric field does the same work;
- The energy changes in O and F decomposed from [O]2− and [F]− in the electrolyte moving to the anode surface and forming Oanode and Fanode are the same as those of [O]2− and [F]− moving to the anode surface and forming Oanode and Fanode.
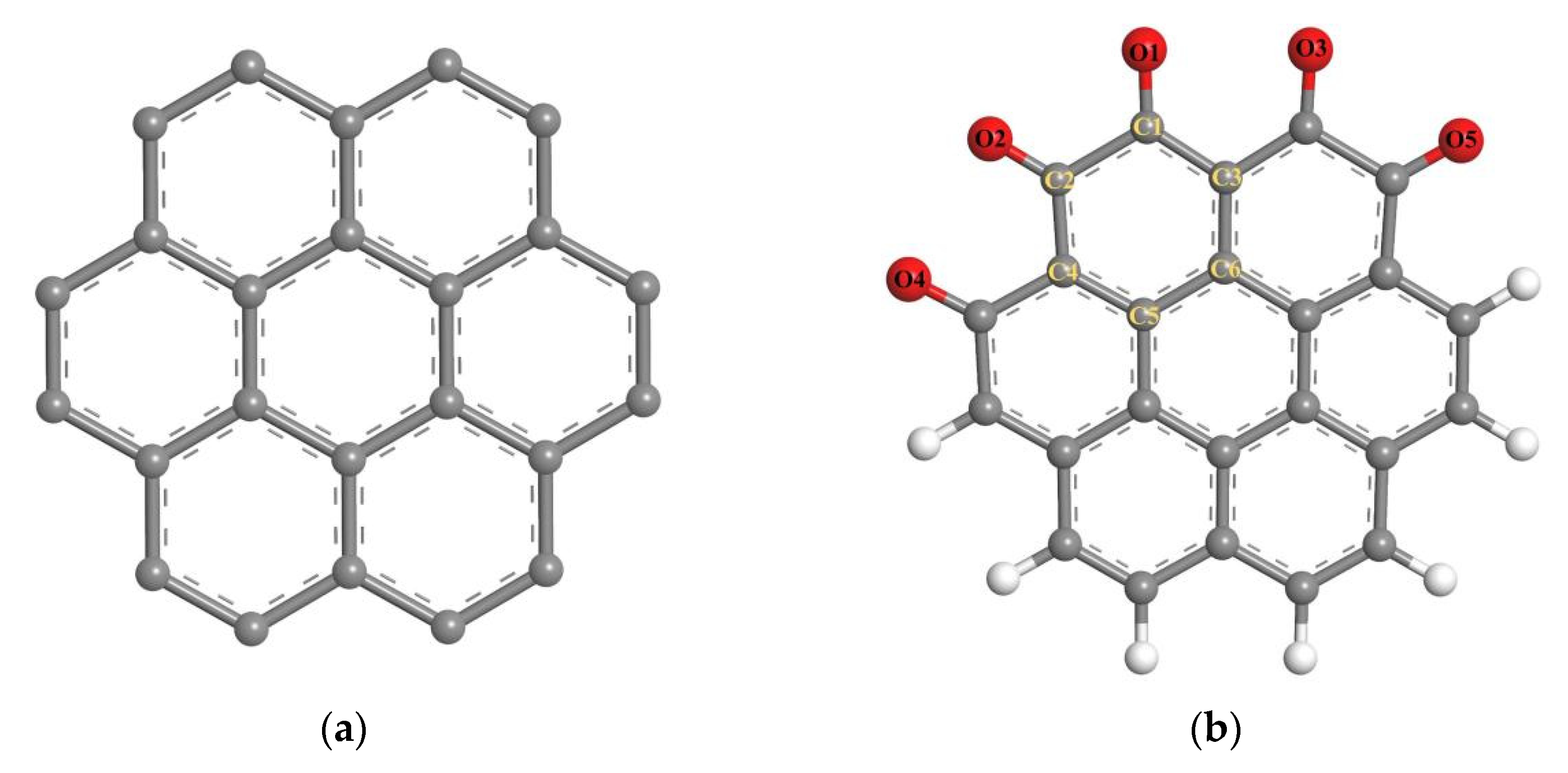
2.1.2. Build Model
2.1.3. Computing Method
2.2. High Temperature Molten Salt Experiment
3. Results and Discussion
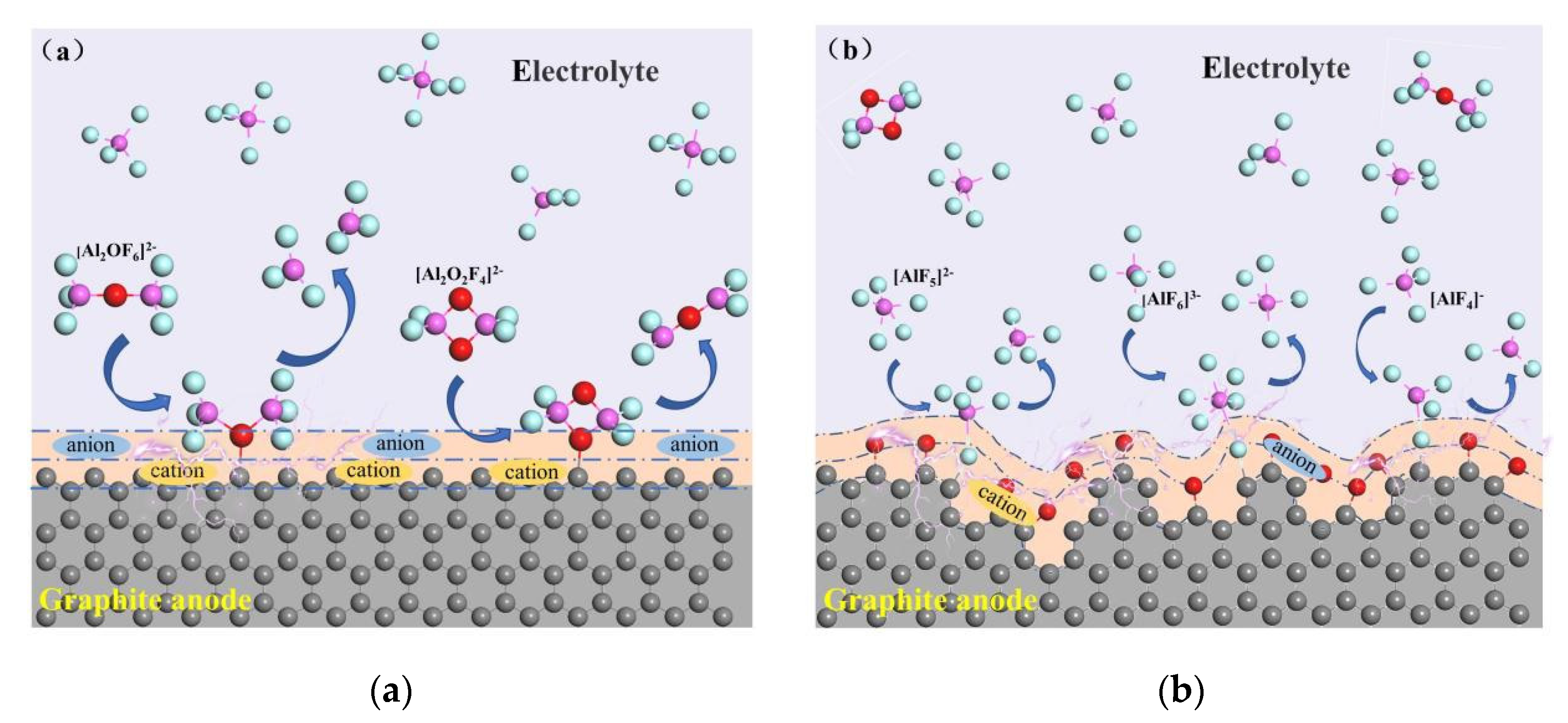
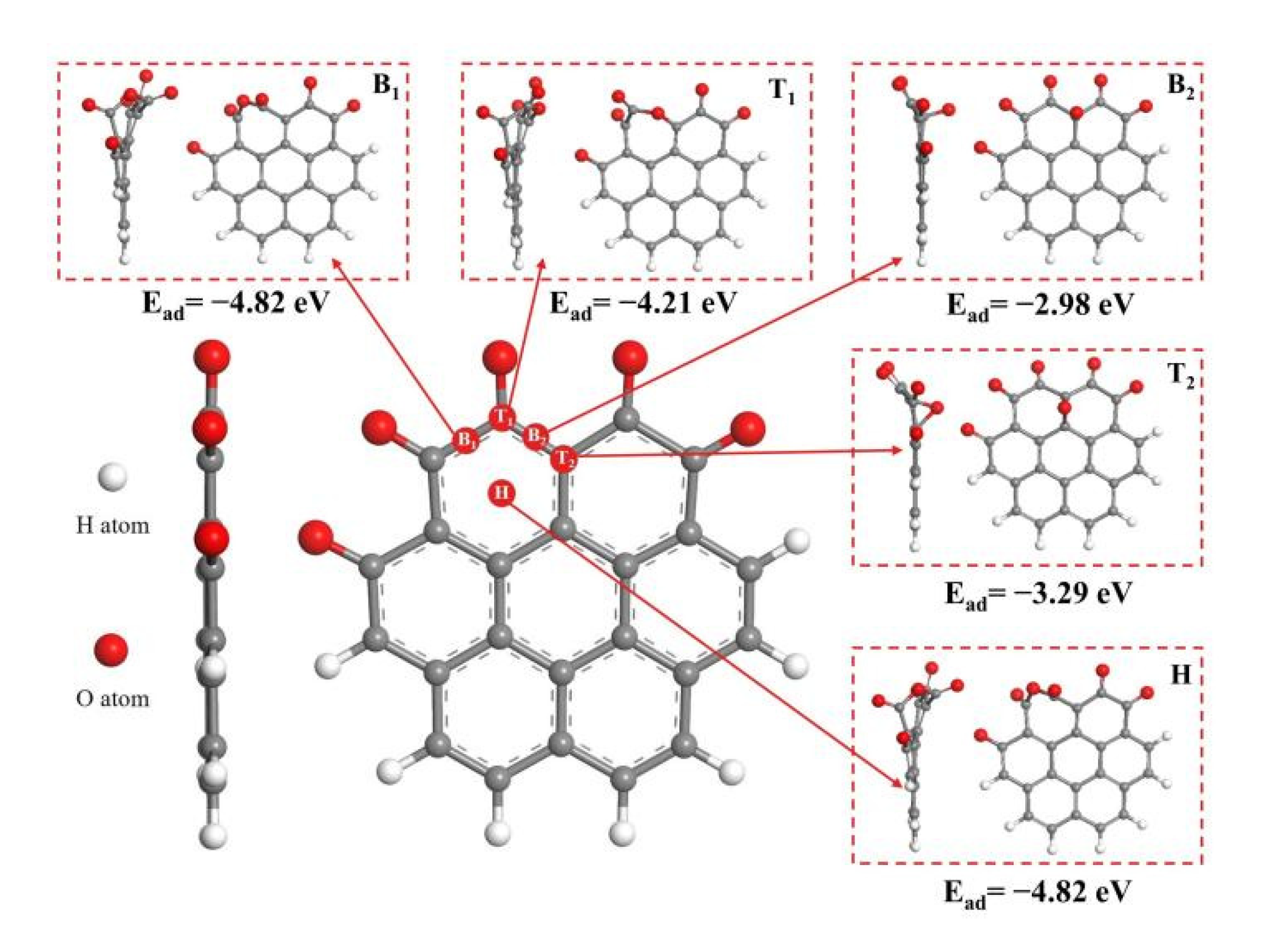
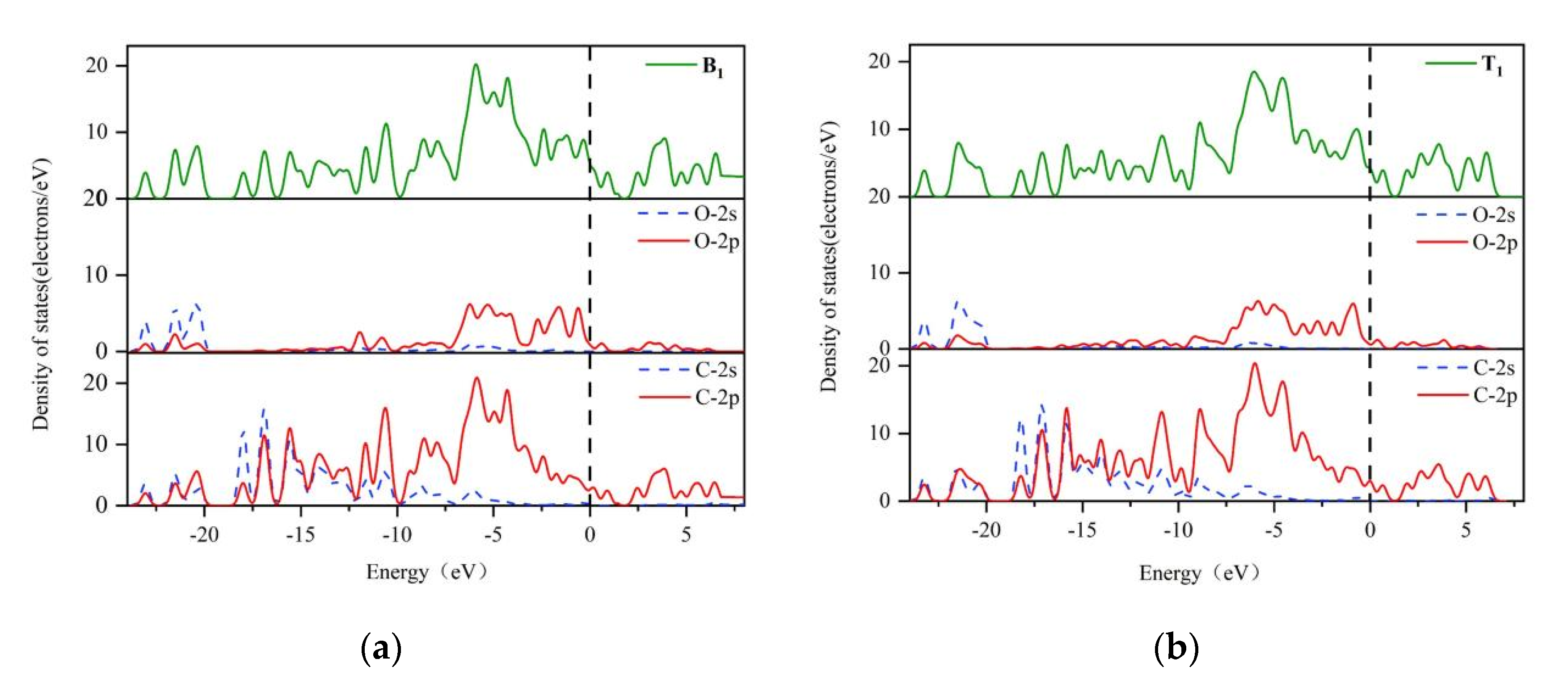
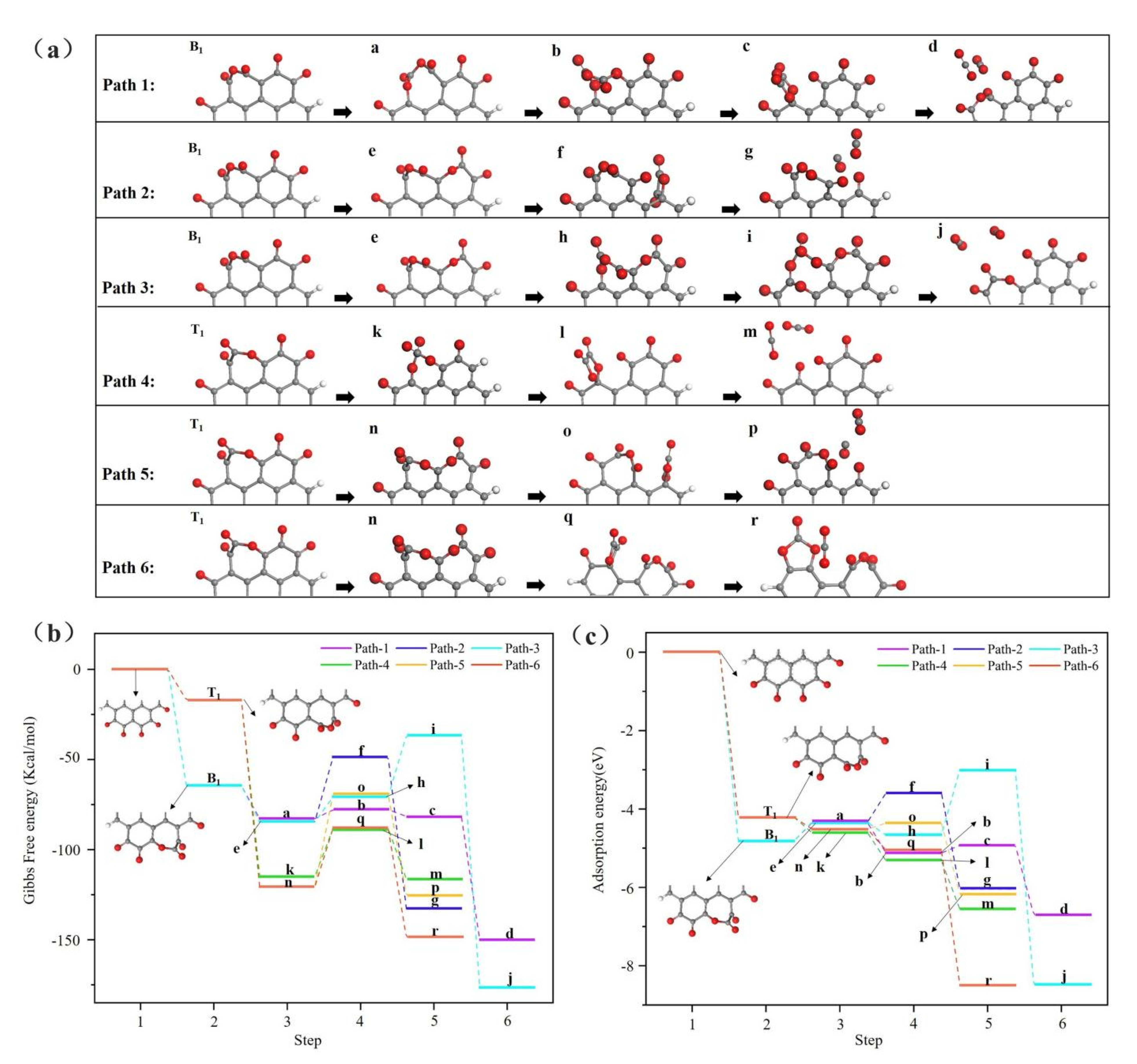
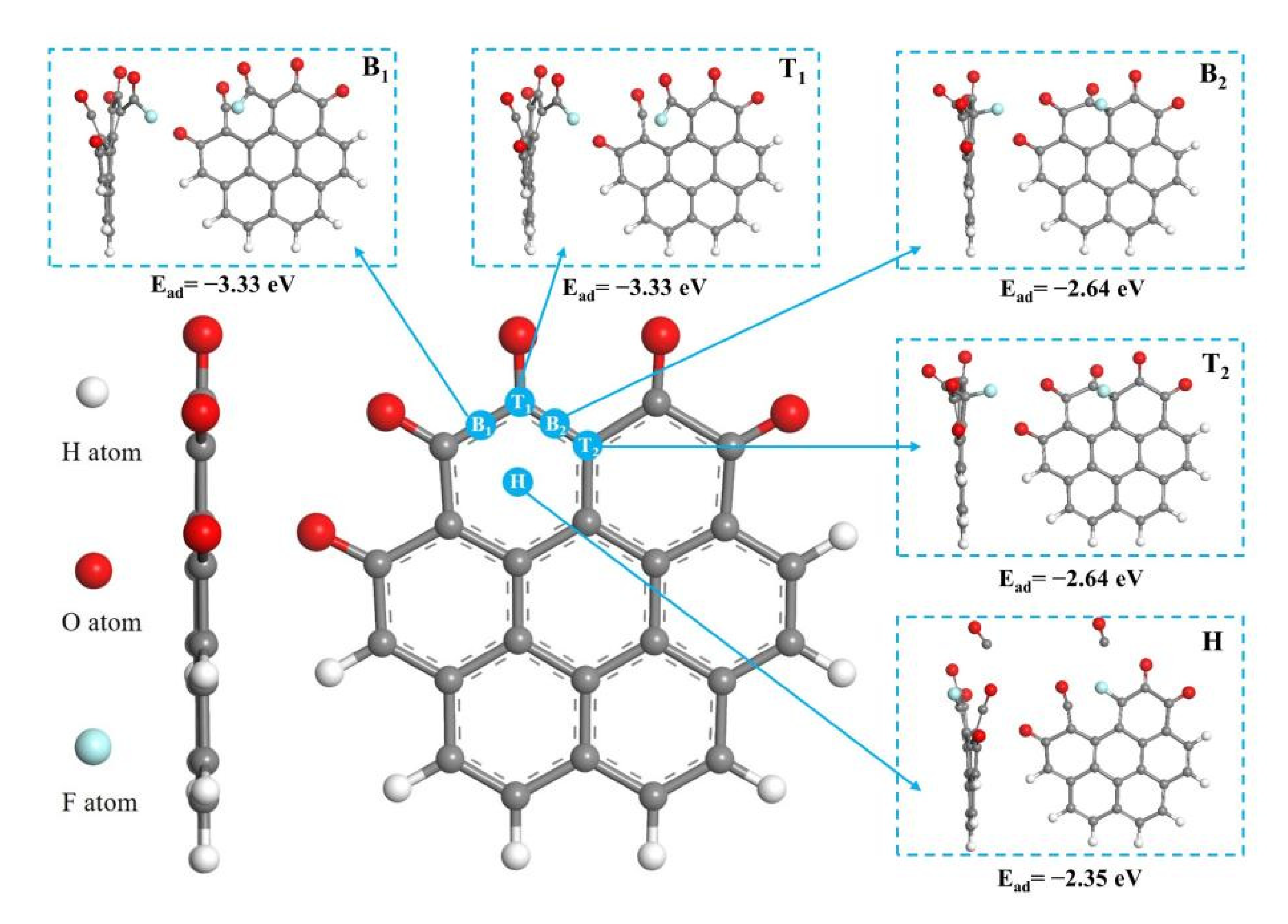
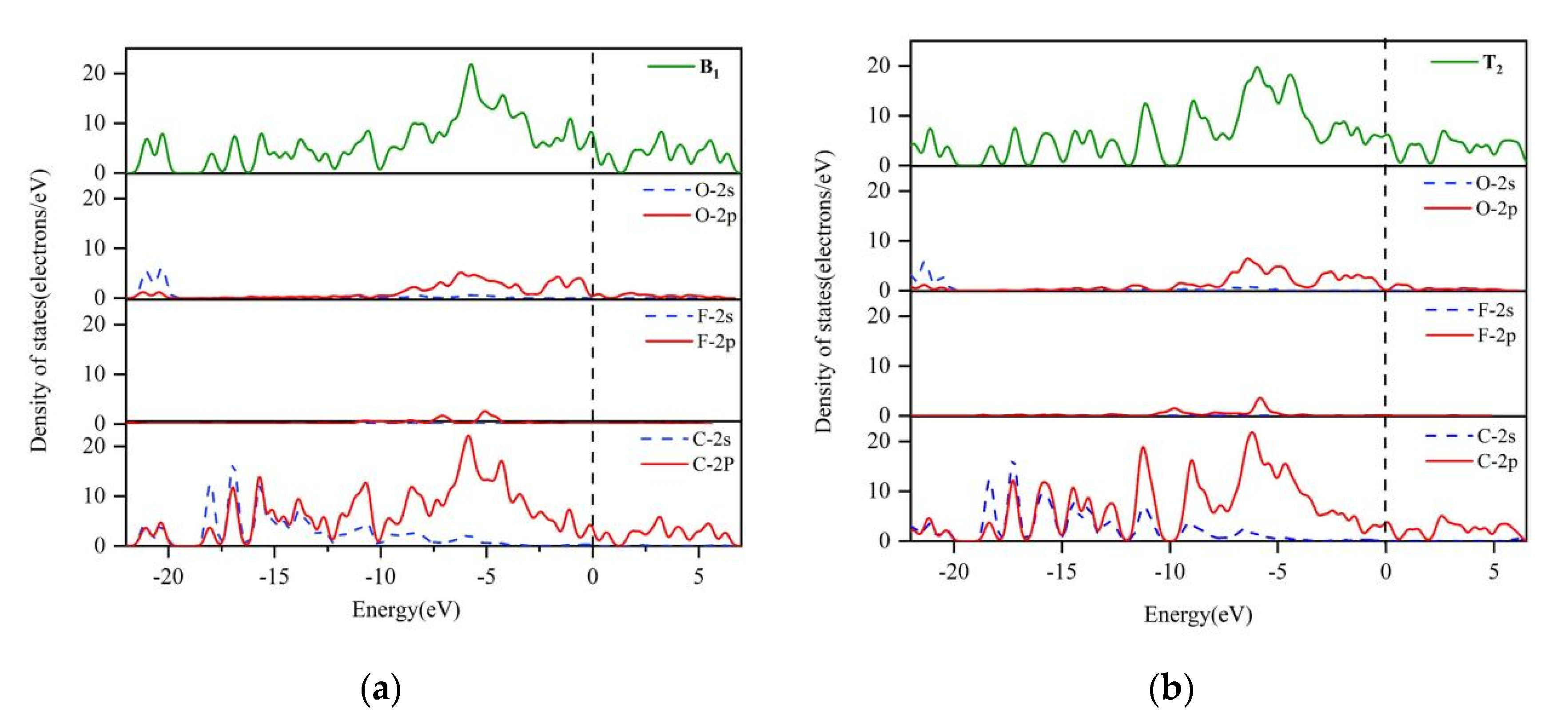
3.1. Reaction at [O]2− Preferential Discharge Stage
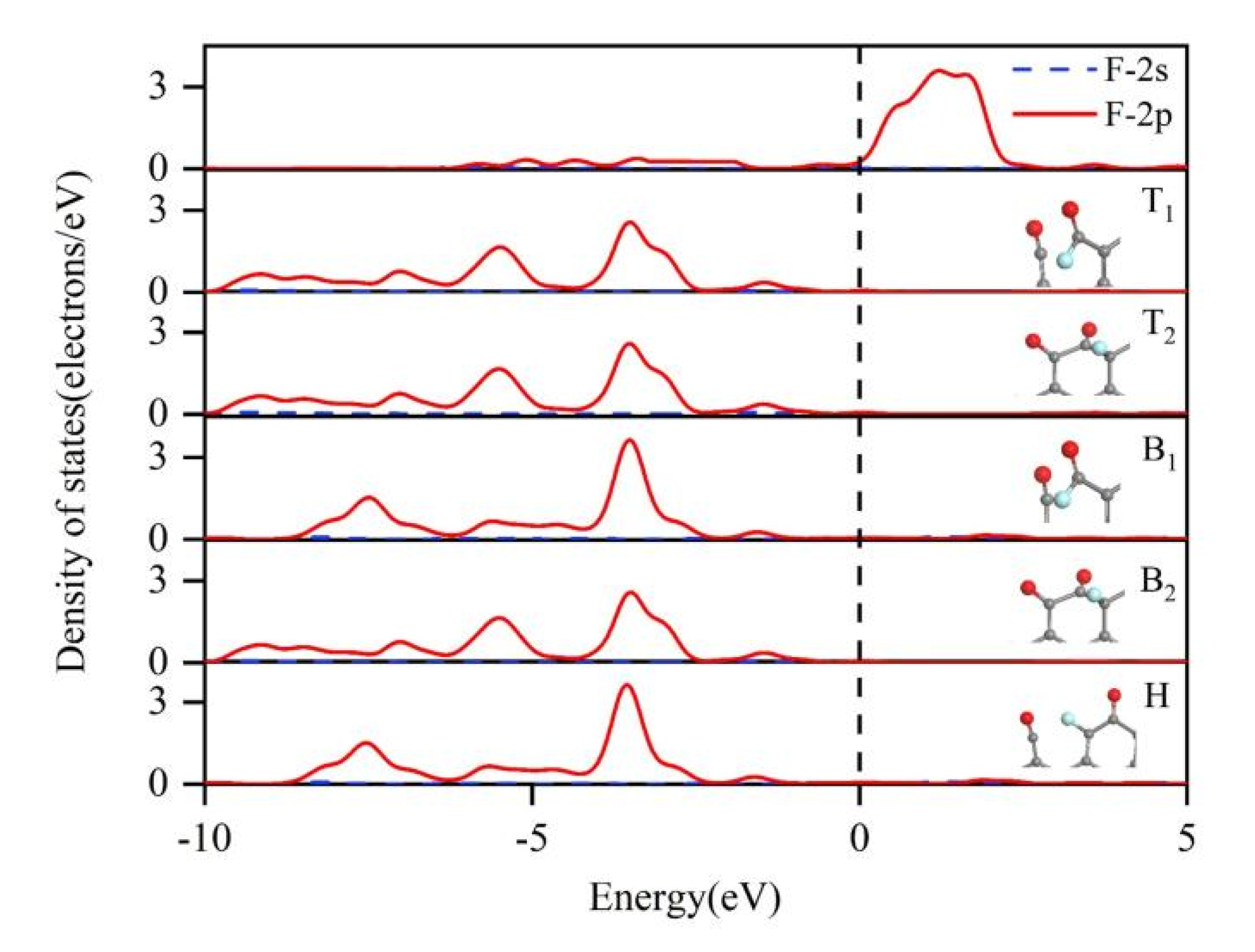
3.2. Reaction at [O]2−, [F]− Co-Discharge Stage
3.3. Molten Salt Experiment Results
4. Conclusions
- 1.
- In the [O]2− preferential discharge stage, the priority of oxygen discharge sites on the anode surface is B1 = H > T1 > T2 > B2. Starting from the B1 and T1 structures, six possible paths for C-O compound generation were calculated, among which Path 3 is the most likely path for CO2 generation;
- 2.
- In the [O]2−, [F]− co-discharge stage, the priority of fluorine ion discharge sites is B1 = T1 > B2 = T2 > H. Starting from the B1 and T2 structures, five paths that possible to generate C-O compounds were calculated, among which the Path 3 is the most likely path for COF2 generation;
- 3.
- The COF2 is an unstable intermediate, which decomposes into CF4 and CO2 under high temperature. This explains why non-anode effect PFC (NAE-PFC) generates a low concentration of CF4;
- 4.
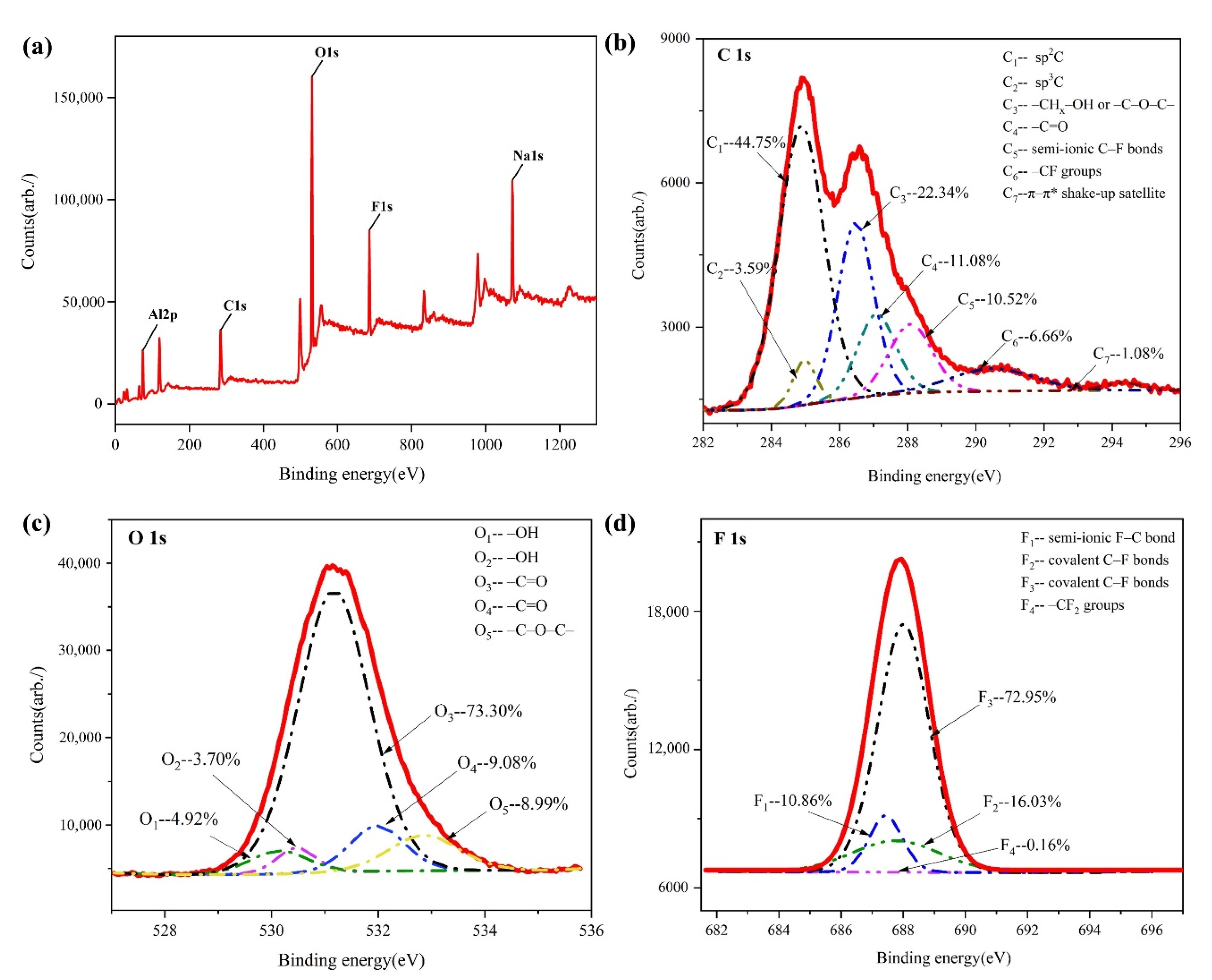
- Experiments proved the existence of -C=O doubles, a -C-O-C- bridge-type structure and -C-F keys on the surface of the anode after electrolysis, indicating that different C-O bonds and C-F bonds are formed during the discharge of O and F. This is consistent with the first-principles calculation.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Saevarsdottir, G.; Kvande, H.; Welch, B.J. Aluminum Production in the Times of Climate Change: The Global Challenge to Reduce the Carbon Footprint and Prevent Carbon Leakage. JOM 2019, 72, 296–308. [Google Scholar] [CrossRef]
- Haraldsson, J.; Johansson, M.T. Effects on primary energy use, greenhouse gas emissions and related costs from improving energy end-use efficiency in the electrolysis in primary aluminium production. Energ. Effic. 2020, 13, 1299–1314. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y.; Zhu, W.; Huang, J. Environmental impact assessment of China’s primary aluminum based on life cycle assessment. T. Nonferr. Metal. Soc. 2019, 29, 1784–1792. [Google Scholar] [CrossRef]
- Gao, F.; Nie, Z.R.; Wang, Z.H.; Hongmei, L.I.; Gong, X.Z.; Zuo, T.Y. Greenhouse gas emissions and reduction potential of primary aluminum production in China. Sci. China Life Sci. 2009, 52, 2161–2166. [Google Scholar] [CrossRef]
- Tabereaux, A.T. Anode effects, PFCs, global warming, and the aluminum industry. JOM 1994, 46, 30–34. [Google Scholar] [CrossRef]
- Mikhaylov, A.; Moiseev, N.; Aleshin, K.; Burkhardt, T. Global climate change and greenhouse effect. Entrep. Sustain. Issues 2020, 7, 2897–2913. [Google Scholar] [CrossRef]
- Kjos, O.S.; Solheim, A.; Aarhaug, T.; Osen, K.S.; Martinez, A.M.; Sommerseth, C.; Gudbrandsen, H.; Store, A.; Gaertner, H. PFC Evolution Characteristics during Aluminium and Rare Earth Electrolysis. In Proceedings of the TMS Light Metals, Phoenix, AZ, USA, 11–15 March 2018; pp. 1449–1455. [Google Scholar]
- Tsai, W.-T.; Chen, H.P.; Hsien, W.Y. A review of uses, environmental hazards and recovery/recycle technologies of perfluorocarbons (PFCs) emissions from the semiconductor manufacturing processes. J. Loss. Prevent. Proc. 2002, 15, 65–75. [Google Scholar] [CrossRef]
- Li, W.X.; Zhao, Q.Y.; Yang, J.H.; Qiu, S.L.; Chen, X.P.; Marks, J.; Bayliss, C. On Continuous PFC Emission Unrelated to Anode Effects. In Proceedings of the TMS Light Metals, San Antonio, TX, USA, 3–7 March 2013; pp. 1037–1045. [Google Scholar]
- Stanic, N.; Bø, E.T.; Sandnes, E. CO and CO2 Anode Gas Concentration at Lower Current Densities in Cryolite Melt. Metals 2020, 10, 1694. [Google Scholar] [CrossRef]
- Harnisch, J.; Borchers, R.; Fabian, P.; Maiss, M. Tropospheric trends for CF4 and C2F6 since 1982 derived from SF6 dated stratospheric air. Geophys. Res. Lett. 1996, 23, 1099–1102. [Google Scholar] [CrossRef]
- Vogel, H.; Flerus, B.; Stoffner, F.; Friedrich, B. Reducing Greenhouse Gas Emission from the Neodymium Oxide Electrolysis. Part I: Analysis of the Anodic Gas Formation. J. Sustain. Metall. 2016, 3, 99–107. [Google Scholar] [CrossRef]
- Heggelund, G.M. China’s climate and energy policy: At a turning point? Int. Environ. Agreem-P. 2021, 21, 9–23. [Google Scholar] [CrossRef]
- Lin, J.Y.; Murshed, M.; Chen, F.Z.; Shahbaz, M.; Kirikkaleli, D.; Khan, Z. An empirical analysis of the household consumption-induced carbon emissions in China. Sustain. Prod. Consump. 2021, 26, 943–957. [Google Scholar]
- Li, S.; Zhang, T.; Niu, L.; Yue, Q. Analysis of the development scenarios and greenhouse gas (GHG) emissions in China’s aluminum industry till 2030. J. Clean. Prod. 2021, 290, 125859. [Google Scholar] [CrossRef]
- Dong, L.; Miao, G.; Wen, W. China’s Carbon Neutrality Policy: Objectives, Impacts and Paths. East. Asian Policy 2021, 13, 5–18. [Google Scholar] [CrossRef]
- Chi, Y.; Liu, Z.; Wang, X.; Zhang, Y.; Wei, F. Provincial CO2 Emission Measurement and Analysis of the Construction Industry under China’s Carbon Neutrality Target. Sustainability 2021, 13, 1876. [Google Scholar] [CrossRef]
- Kuang, Z.; Thonstad, J.; Sørlie, M. Effects of additives on the electrolytic consumption of carbon anodes in aluminium electrolysis. Carbon 1995, 33, 1479–1484. [Google Scholar] [CrossRef]
- Dorreen, M.M.R.; Hyland, M.M.; Welch, B.J. Current Efficiency Studies in a Laboratory Aluminium Cell Using the Oxygen Balance Method. In Proceedings of the TMS Light Metals, San Antonio, TX, USA, 3–7 March 2013; pp. 204–210. [Google Scholar]
- Liu, D.; Li, X.; Bai, Y. An attack-finding algorithm for security protocols. J. Comput. Sci. Tech. 2002, 17, 450–463. [Google Scholar] [CrossRef]
- Huglen, R.; Kvande, H. Global Considerations of Aluminium Electrolysis on Energy and the Environment. In Proceedings of the TMS Light Metals, Phoenix, AZ, USA, 11–15 March 2018; pp. 948–955. [Google Scholar]
- Wang, G.; Sun, X.; Zhao, W.; Yang, D. Electrochemical Behaviour of Carbon Anodes in Na3AlF6-K3AlF6-Based Low-Melting Electrolytes for Aluminium Electrolysis. In Proceedings of the TMS Light Metals, Nashville, TN, USA, 14–18 February 2016; pp. 671–674. [Google Scholar]
- Åsheim, H.; Aarhaug, T.A.; Sandnes, E.; Kjos, O.S.; Solheim, A.; Kolås, S.; Haarberg, G.M. Anode Effect Initiation during Aluminium Electrolysis in a Two-Compartment Laboratory Cell. In Proceedings of the TMS Light Metals, Phoenix, TN, USA, 14–18 February 2016; pp. 551–556. [Google Scholar]
- Yang, Y.; Gao, B.; Wang, Z.; Shi, Z.; Hu, X. Study on the Inter-electrode Process of Aluminum Electrolysis. Metall. Mater. Trans. B 2015, 47, 621–629. [Google Scholar] [CrossRef]
- Xiao, J.; Li, J.; Ye, S.-l.; Lai, Y.-q.; Liu, Y.-x. Laboratory study of property-modified prebaked carbon anode and application in large aluminum electrolysis cells. J. Cent. South Univ. T. 2005, 12, 68–71. [Google Scholar]
- Wrigge, F.W.; Ginsberg, H. Zur Kenntnis der Vorgange beim Ausruhren technischen Tonerdehydrates. Z. Anorg. Allg. Chem. 1951, 264, 285–297. [Google Scholar] [CrossRef]
- Thonstad, J.; Nordmo, F.; Rødseth, J.K. On the anode effect in cryolite-alumina melts—II the initiation of the anode effect. Electrochim. Acta. 1974, 19, 761–769. [Google Scholar] [CrossRef]
- Nordmo, F.; Thonstad, J. On the anode effect in cryolite-alumina melts—IV. Gas composition and faradaic efficiency. Electrochim. Acta. 1985, 30, 741–745. [Google Scholar] [CrossRef]
- Dion, L.; Kiss, L.I.; Poncsák, S.; Lagacé, C.-L. Prediction of Low-Voltage Tetrafluoromethane Emissions Based on the Operating Conditions of an Aluminium Electrolysis Cell. JOM 2016, 68, 2472–2482. [Google Scholar] [CrossRef] [Green Version]
- Hyland, M. Non Anode Effect PFCS: Measurement Considerations and Potential Impacts. In Proceedings of the TMS Light Metals, Orlando, FL, USA, 15–19 March 2015; pp. 551–555. [Google Scholar]
- Dorreen, M.M.R.; Chin, D.L.; Lee, J.K.C.; Hyland, M.M.; Welch, B.J. Sulfur and Fluorine Containing Anode Gases Produced during Normal Electrolysis and Approaching an Anode Effect. In Proceedings of the TMS Light Metals, San Antonio, TX, USA, 3–7 March 2013; pp. 918–923. [Google Scholar]
- Jarek, S.; Thonstad, J. Fast voltammetry study of anodic reactions in graphite in cryolite-alumina melts. Electrochim. Acta. 1987, 32, 743–747. [Google Scholar] [CrossRef]
- Calandra, A.J.; Castellano, C.E.; Ferro, C.M. The electrochemical behaviour of different graphite/cryolite alumina melt interfaces under potentiodynamic perturbations. Electrochim. Acta. 1979, 24, 425–437. [Google Scholar] [CrossRef]
- Djokić, S.S.; Conway, B.E.; Belliveau, T.F. Specificity of anodic processes in cyclic voltammetry to the type of carbon used in electrolysis of cryolite-alumina melts. J. Appl. Electrochem. 1994, 24, 827–834. [Google Scholar] [CrossRef]
- Lei, Y.; Shevlin, S.A.; Zhu, W.; Guo, Z.X. Hydrogen-induced magnetization and tunable hydrogen storage in graphitic structures. Phys. Rev. B 2008, 77, 761–768. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Yu, G.; Hou, G.; Qiu, S. Study on the identification of PFC emission from aluminum reduction cells. Light Metals 2017, 11, 11–39. [Google Scholar]
- Bou, M.; Martin, J.M.; Le Mogne, T.; Vovelle, L. Chemistry of the interface between aluminium and polyethyleneterephthalate by XPS. Appl. Surf. Sci. 1991, 47, 149–161. [Google Scholar] [CrossRef]
- Weng, L.T.; Poleunis, C.; Bertrand, P.; Carlier, V.; Sclavons, M.; Franquinet, P.; Legras, R. Sizing removal and functionalization of the carbon fiber surface studied by combined TOF SIMS and XPS. J. Adhes. Sci. Technol. 1995, 9, 859–871. [Google Scholar] [CrossRef]
- Endo, K.; Inoue, C.; Kobayashi, N.; Aida, M. Spectra analysis of the XPS core and valence energy levels of polymers by an ab initio mo method using simple model molecules. J. Phys. Chem. Solids 1994, 55, 471–478. [Google Scholar] [CrossRef]
- López, G.P.; Castner, D.G.; Ratner, B.D. XPS O 1s binding energies for polymers containing hydroxyl, ether, ketone and ester groups. Surf. Interface Anal. 1991, 17, 267–272. [Google Scholar] [CrossRef]
- Groult, H.; Lantelme, F.; Crassous, I.; Labrugere, C.; Tressaud, A.; Belhomme, C.; Colisson, A.; Morel, B. Characteristics of the Fluorocarbon Surface Film Generated on Carbon Anode during F2 Evolution in Molten KF-2HF. J. Electrochem. Soc. 2007, 154, 553–562. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.; Li, J.; Zhang, H.; Li, T.; Li, J. First-Principles Study on the Mechanism of Greenhouse Gas Generation in Aluminum Electrolysis. Metals 2021, 11, 1118. https://doi.org/10.3390/met11071118
Sun K, Li J, Zhang H, Li T, Li J. First-Principles Study on the Mechanism of Greenhouse Gas Generation in Aluminum Electrolysis. Metals. 2021; 11(7):1118. https://doi.org/10.3390/met11071118
Chicago/Turabian StyleSun, Kena, Jie Li, Hongliang Zhang, Tianshuang Li, and Jiaqi Li. 2021. "First-Principles Study on the Mechanism of Greenhouse Gas Generation in Aluminum Electrolysis" Metals 11, no. 7: 1118. https://doi.org/10.3390/met11071118
APA StyleSun, K., Li, J., Zhang, H., Li, T., & Li, J. (2021). First-Principles Study on the Mechanism of Greenhouse Gas Generation in Aluminum Electrolysis. Metals, 11(7), 1118. https://doi.org/10.3390/met11071118