3.1. Vanadium Dissolution and Deposition in Equimolar AlCl3 + NaCl Melt
To analyse the anodic and cathodic processes taking part on the vanadium electrode in equimolar AlCl
3 + NaCl melt, potentiodynamic polarization curve and cyclic voltammetry measurements were performed,
Figure 1 and
Figure 2.
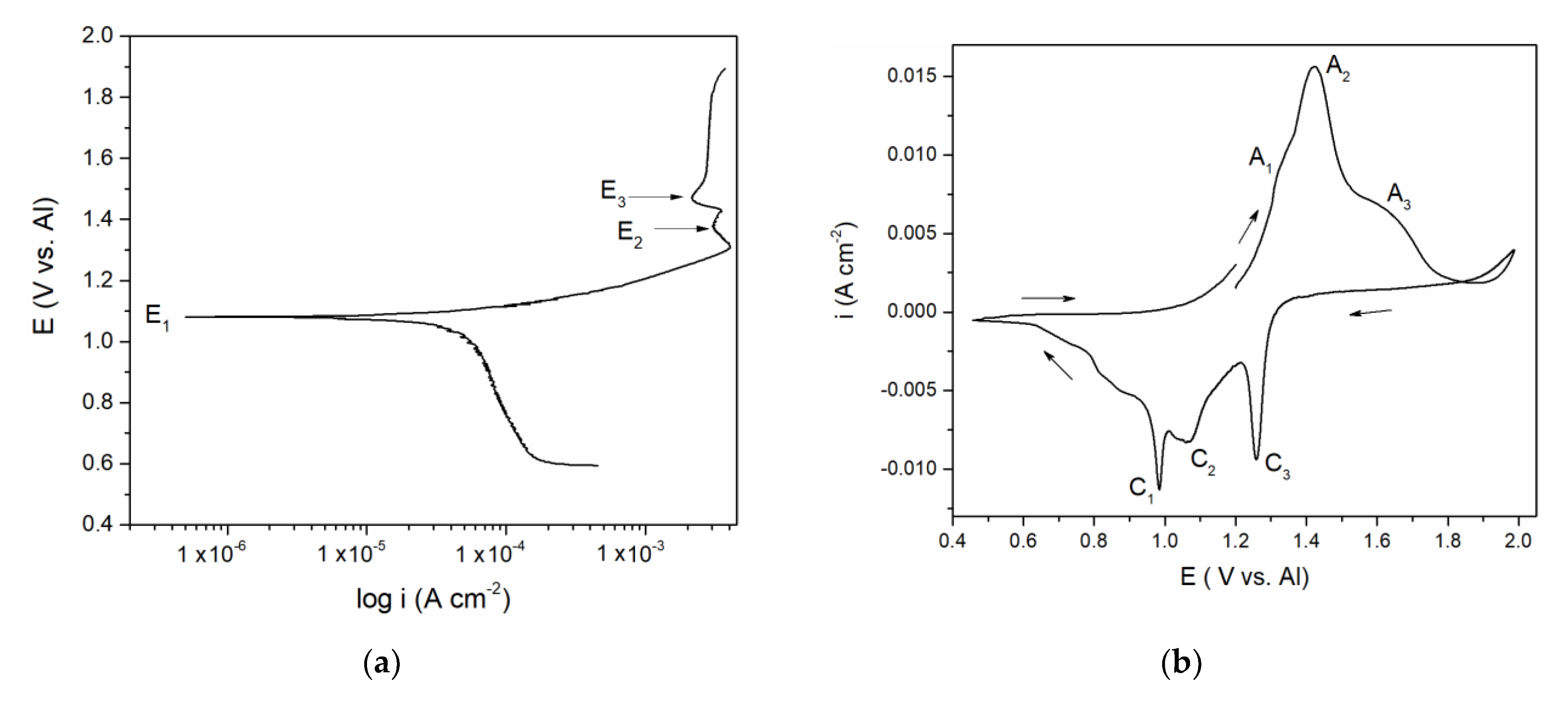
Polarization curve in
Figure 1a suggests a well-defined potential E
1 at 1.100 V vs. Al and less pronounced potentials E
2 at ≈ 1.350 and E
3 at ≈ 1.480 V vs. Al which correspond rather well with the “calculated equilibrium potentials” defined as a midpoint values between anodic and cathodic peak pairs (A
1 + C
1)/2 = 1.150 V, (A
2 + C
2)/2 = 1.350 V and (A
3 + C
3)/2 = 1.500 V vs. Al from
Figure 1b. The recorded calculated and peak potentials were more than one volt anodic to the equilibrium potential of aluminium in the electrolyte used and therefore had to be attributed to the reversible reactions of the remaining constituents of the electrolyte. Chlorine evolution was found to start at potentials more positive than 2.000 V vs. Al and it is known that sodium equilibrium potential in chloride melts is substantially more negative than that of aluminium [
15,
16]. The recorded equilibrium and peak potentials, therefore, had to be attributed to vanadium species in metallic and ionized forms.
Vanadium ions were added to the equimolar electrolyte by dissolution of the vanadium working electrode. There are several studies on the vanadium ions being present in the chloride melts (NaCl + KCl at 690 to 860 °C) [
15,
16]. The equilibrium potentials of the reactions: V(II) ↔ V(0), V(III) ↔ V(II), Cl
− + 2e
− ↔ Cl
2, were established [
15,
16]. In our study, the results suggested that in the presence of aluminium ions in equimolar chloroaluminate electrolyte (0.19 mol AlCl
3 + 0.19 mol NaCl at 200 to 300 °C) vanadium behaved very similarly. This helped us to attribute the recorded potentials E
1 ≈ 1.100 V, E
2 ≈ 1.350 V and E
3 ≈ 1.480 V vs. Al in
Figure 1a to V(II) ↔ V(0), V(III) ↔ V(II), and V(IV) ↔ V(III) reactions, respectively. It was then possible to identify the processes reflected by the current waves in
Figure 1b: current wave A
1, should be related to the process of vanadium dissolution V(0) → V(II); A
2 at more anodic potentials presents further oxidation reaction V(II) → V(III); current wave A
3, at even more positive potentials, reflects the oxidation process of V(III) → V(IV),
Figure 1b. In the reverse scan, C
3 and C
2 are reflecting the reduction processes of vanadium ions V(IV) → V(III), and V(III) → V(II), respectively. Current peak C
1 reflects the reduction of V(II) ions to the metal vanadium and the deposition onto the vanadium working electrode.
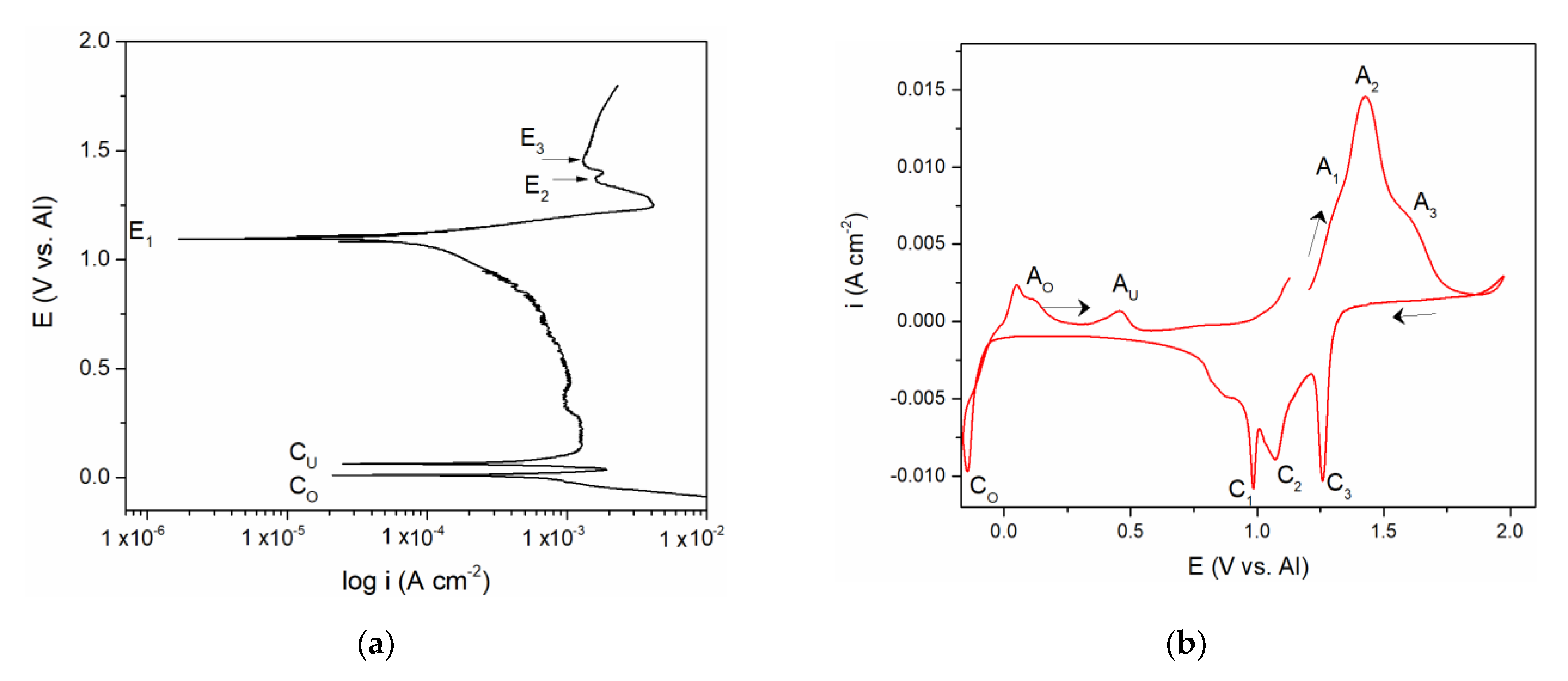
The polarization curve and cyclic voltammograms recorded on the vanadium working electrode which included aluminium deposition and dissolution in addition to the vanadium dissolution and deposition are presented in
Figure 2a,b. Polarization curve,
Figure 2a, starting from the potentials negative to aluminium equilibrium potential in the electrolyte used revealed two well-defined potentials, C
O at ≈ 0.000 V and C
U at ≈ 0.080 V vs. Al, and a poorly defined one at C
UP ≈ 0.350 V vs. Al, followed by the potentials E
1, E
2, and E
3. It was observed that the values of the three most positive potentials (E
1, E
2, and E
3) are identical to those three recorded by polarization curve in
Figure 1a and could be attributed to the same reactions of vanadium dissolution/deposition and vanadium ion oxidation/reduction.
CV measurements presented in
Figure 2b, were done as follows:
- -
Starting potential was 1.200 V vs. Al, the potential more positive to the vanadium equilibrium potential in the equimolar AlCl3 + NaCl electrolyte;
- -
Potential was then changed to 2.000 V vs. Al, which is close enough to the chlorine evolution potential;
- -
At 2.000 V vs. Al, the potential was then directed toward cathodic end value (ECF = –0.150 V vs. Al) which was cathodic to the Al equilibrium potential in the used electrolyte;
- -
Potential was then returned to the starting value (1.200 V vs. Al).
Tracing the path of the system response recorded on the voltammogram, we first see three anodic current peaks (A
1, A
2, and A
3) and their three cathodic counterparts (C
1, C
2, and C
3) in the region which is defined between 0.600 and 2.000 V vs. Al in the graph. The values of the peak potentials were very similar to those presented in
Figure 1b and attributed to the same processes of vanadium dissolution/deposition and oxidation/reduction of the vanadium ions.
The values of the peak potentials related to the current waves (A
1, A
2, A
3, and C
1, C
2, C
3) and their peaks,
Figure 2b, were in very good accordance with the values of the three most anodic calculated equilibrium potentials presented by the potentiodynamic polarization curve of the vanadium working electrode,
Figure 2a. These data were also in agreement with the conclusions obtained by Polovov at al. describing vanadium dissolution in sodium-potassium chloride melts, at temperatures above 680 °C [
16].
To investigate further the processes of vanadium dissolution/deposition and vanadium ion oxidation/reduction that are represented by A
1/C
1 and A
2/C
2 current waves in
Figure 1 and
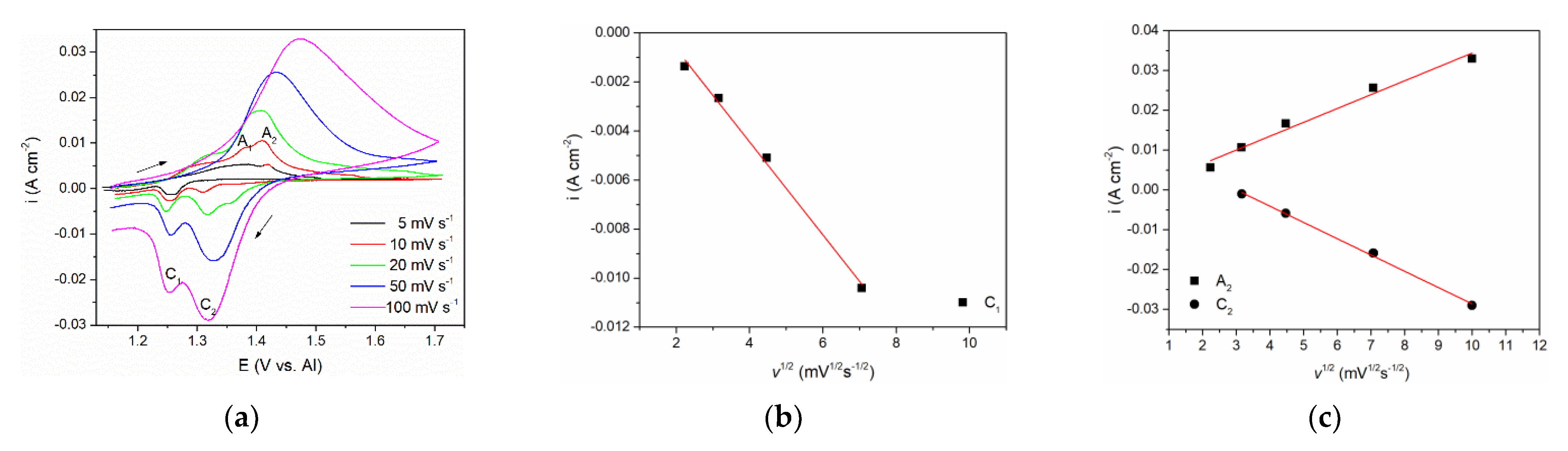
Figure 2, a series of CVs spanning their potential range with scan rates from 5 mVs
−1 to 100 mVs
−1 were performed, see
Figure 3a. The relationships between the peak maximum current densities and the square root of the scan rate applied are presented in
Figure 3b,c.
The CVs representing lower scan rates (5 and 10 mVs
−1) showed two distinct peak potentials A
1 and A
2 in the anodic section of the voltammograms. Peak A
1 was attributed to the dissolution process of vanadium metal: V(0) → V(II), and peak A
2 reflects further oxidation of divalent vanadium (II) i.e., V(II) → V(III). With increased scan rate, only one broader peak was observed, occupying the potential range of both A
1 and A
2. This behavior is typically a result of the next process (current wave A
2) starting at an increasing rate while the previous process (current wave A
1) rate is fading away [
16]. Thus, the obtained one united current wave represents the sum of rates of both processes taking part in a partly shared potential span. Therefore, a proper analysis i
peak = f(
v1/2) for A
1 could not be made. However, analysis of its cathodic counterpart, the deposition current wave C
1, was possible and showed that the peak current vs. square root of the scan rate is linear and does not pass through the origin. This implies that the V(II) ion reduction process V(II) → V(0) is under mixed control: by the diffusion rate of the complexed vanadium ions from the bulk of the molten equimolar AlCl
3 + NaCl electrolyte toward the WE and by the rate of the charge transfer step [
23]. Some other authors also came to the conclusion that both anodic dissolution of vanadium and vanadium deposition in sodium-potassium chloride melts are under the same mixed control [
15,
16,
24].
With the scan rate increased, maximum current densities for both oxidation (A
2) and reduction peaks (C
2), were higher, but their peak potentials barely changed implying the reversibility of the redox process V(III) ↔ V(II). Analysis of the relationship i
peak = f(
v1/2) for both A
2 and C
2 presented in
Figure 3c revealed linear relationships suggesting that oxidation of V(II) ions into V(III) ions and the reverse reduction V(III) → V(II) processes are under mixed mass transport and charge transfer control, as suggested earlier in the literature in somewhat different electrolytes [
15,
16,
24].
These findings were used in defining the regime for vanadium metal dissolution as a supply of vanadium ions to the equimolar AlCl
3 + NaCl molten salt electrolyte. Taking into account the results obtained by CV on the vanadium WE in the equimolar electrolyte system (
Figure 1,
Figure 2 and
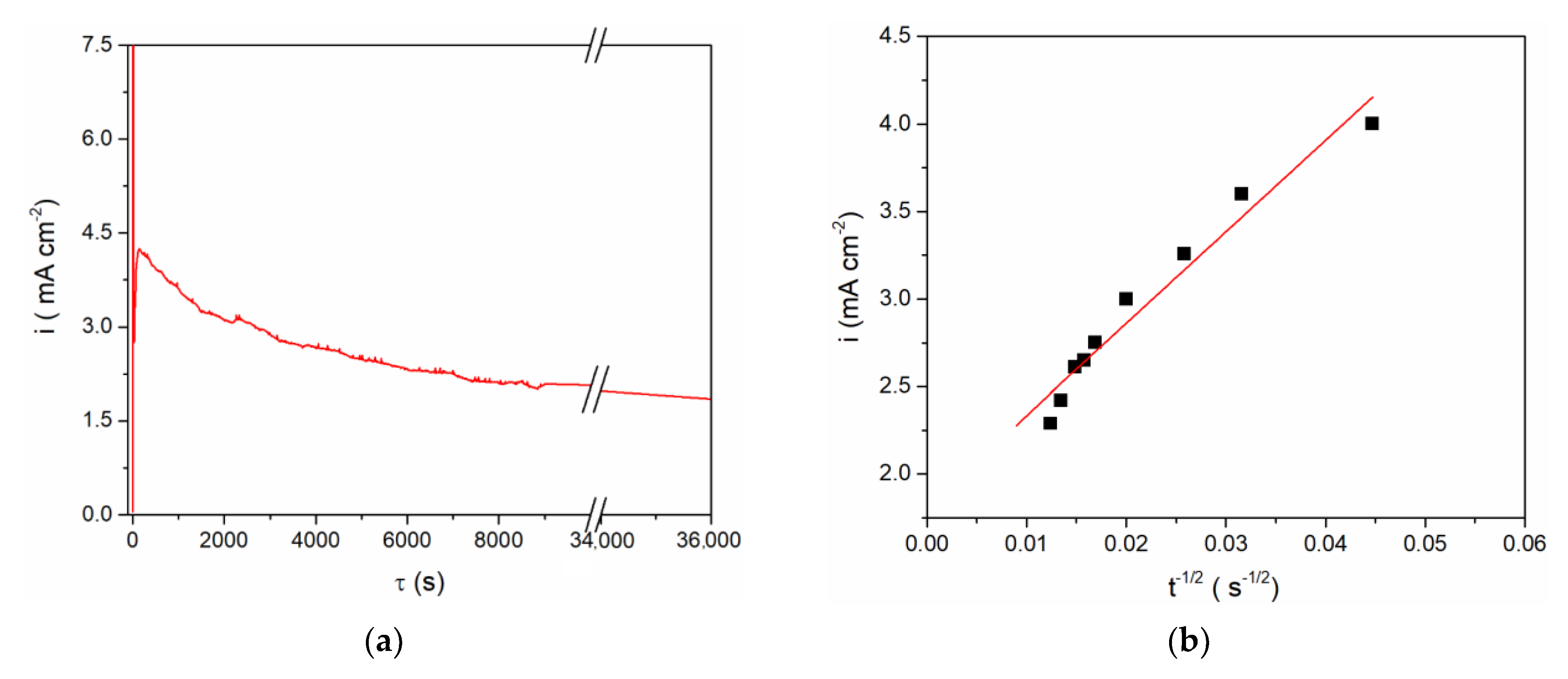
Figure 3), controlled anodic dissolution of vanadium was done by applying a constant potential of 1.300 V vs. Al at 200 °C. The chosen potential was anodic to the vanadium OCP,
Figure 4, and anodic enough to sustain vanadium metal dissolution. However, analysis of the current density-time diagram in
Figure 4a expressed as the i = f(t
−1/2) relationship appeared to be approximately linear,
Figure 4b, thus identifying the dissolution as predominantly mass transfer controlled [
23].
The final concentration of vanadium ions in the electrolyte obtained by anodic vanadium dissolution was determined in two ways: (1) from Faraday’s Law, using the number of exchanged electrons as n = 2 with respect to the oxidation process, V(0) → V(II), for example
Figure 4a; and (2) from the vanadium working electrode mass lost during controlled potentiostatic dissolution. The results obtained by these two methods agreed well and showed that after controlled electro-dissolution the vanadium (II) ions concentration in the equimolar AlCl
3 + NaCl electrolyte was ≈ 5 millimolar.
3.2. Vanadium and Aluminium Deposition from Equimolar AlCl3 + NaCl Melt Containing V Ions Introduced Either by Anodic Dissolution of V Metal or by Adding VCl3
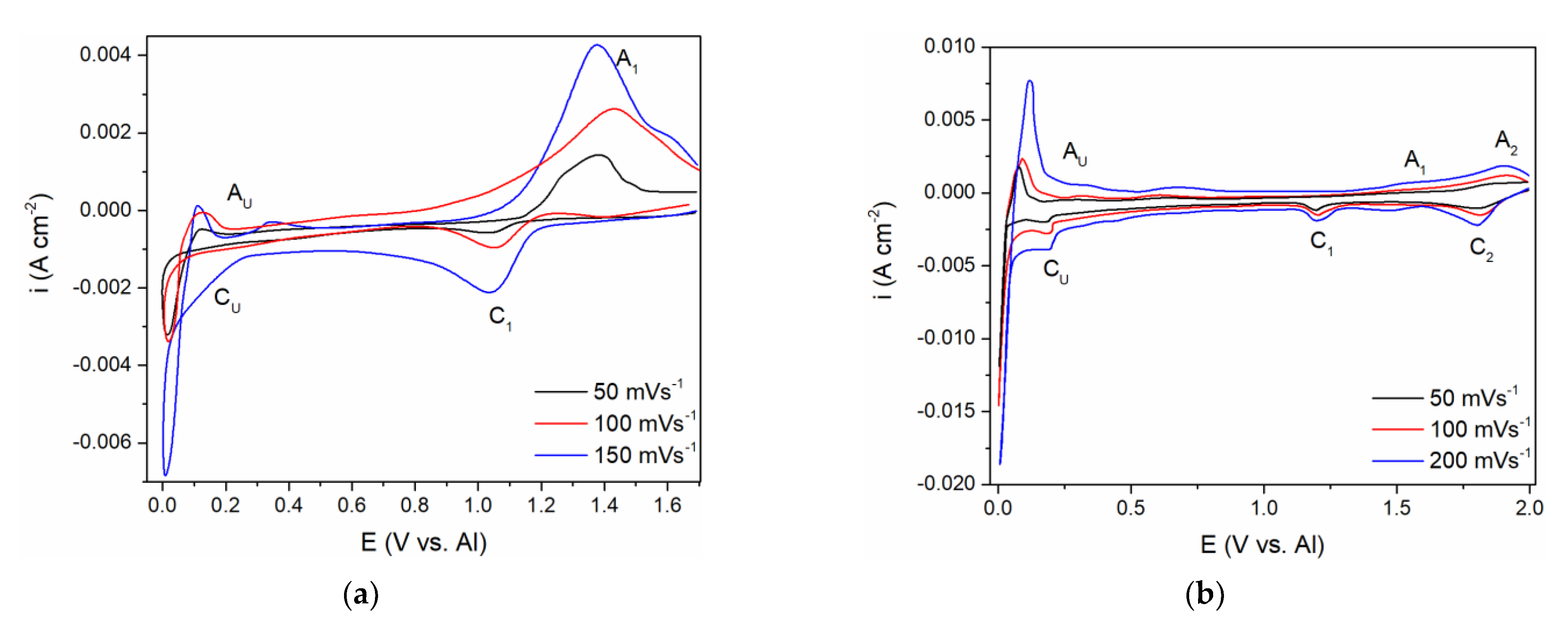
A comparative study of the CV measurements was conducted with the GC working electrode in (a) the equimolar melt, which was 5 millimolar with respect to the vanadium ions introduced by vanadium anode dissolution, and (b) the equimolar melt, which was made to be 12.5 millimolar with respect to the vanadium ions by added VCl
3. The WE potential was scanned from E
I values positive to the vanadium equilibrium potential to E
CF close to the aluminium equilibrium potential and back, enabling the recording of the ongoing electrochemical reactions related to the vanadium and aluminium in the chosen electrolytes,
Figure 5a,b.
From the voltammogram obtained in the equimolar melt containing anodically dissolved V ions, the cathodic peak (C
1) at around 1.000 V vs. Al in the scanned potential region is reflecting V(II) → V(0) ions reduction into vanadium metal,
Figure 5a, and the equilibrium potential of V(II) ↔ V(0) was noted at ≈1.100 V vs. Al. The structure of the anodic and cathodic peaks appearing in the potential range from 1.000 V to 2.000 V vs. Al in both systems on GC presented in
Figure 5a,b is not as detailed as on the V electrode (see
Figure 1b,
Figure 2b and
Figure 3a). Recent studies in chloride melts indicated that anodic dissolution of vanadium metal produces V(II) species, and the presence of vanadium metal in the electrolyte system suppresses the disproportion of V(II) [
16,
25]. According to the results obtained in our study, (
Figure 5), it seems that vanadium ions added to the equimolar AlCl
3 + NaCl melt by controlled electrochemical dissolution behave in the same manner.
Current from the wave of vanadium deposition C
1 in
Figure 5a,b, towards more negative potentials, diminishes but remains at the value which can be recognized as equal or close to the diffusion controlled, vanadium deposition limiting current.
At potentials from 0.200 to 0.300 V positive to the aluminium equilibrium potential, reduction current rises again into a visible wave C
U. Its value cannot possibly reflect just the vanadium diffusion limited deposition current any more but might reflect a sum of the underpotential deposition (UPD) of aluminium onto vanadium previously deposited on the original working electrode and the vanadium diffusion limited deposition current [
8,
9].
The voltammograms recorded in the equimolar melt with VCl
3 added, under all other conditions kept the same, presented a different number of reduction current peaks in the potential range from 2.000 to 1.000 V vs. Al,
Figure 5b. Two distinct cathodic peak potentials were recognized:
Peak C1 at ≈ 1.800 V vs. Al, which should reflect the reduction process of V(III) + e− → V(II)
Peak C2 at ≈ 1.180 V vs. Al, which should depict the deposition process of V(II) + 2e− → V(0).
The presence of the two reduction waves (C
1 and C
2) within the potential range from 2.000 to 1.000 V vs. Al is in accordance with the previous findings that VCl
3 dissolved in chloride melts yields V(III) ions [
15].
The appearance of the reduction current wave C
U at around 0.200–0.300 V vs. Al as a follow-up to the vanadium diffusion-controlled deposition current in
Figure 5b, indicates aluminium underpotential deposition (UPD) onto metal vanadium covering the surface of the original WE. This was recognized in our previous works, and recorded as C
UP in
Figure 2a [
8,
9]. Further increase in the current density, within the C
U current wave should be the result of summing up of the vanadium mass controlled deposition current and aluminium underpotential deposition current growing with increased negativity of the WE potential,
Figure 5a,b. It should be noted that the diffusion-controlled vanadium deposition current in the potential range between ≈ 1.100 V to ≈ 0.250 V vs. Al, in the electrolyte made with VCl
3 (
Figure 5b), is 2–3 times higher than in the electrolyte with anodic dissolution obtained vanadium ions (
Figure 5a). It is to be expected because the V(II) concentration in the first electrolyte was 2.5 to 3 times higher than in the latter.
The current waves, A
1 in
Figure 5a; A
1 and A
2 in
Figure 5b; A
1, A
2, A
3 in
Figure 2b, are oxidation counterparts to the cathodic reactions C
1, C
2, and C
3. In all three electrolytes used, A
1, as a response to C
1, reflects vanadium dissolution while A
2 and A
3 reflect the oxidation part of the redox reaction between V(II), V(III), and V(IV) ions.
The second group of anodic current waves, a number of current waves marked as A
U in
Figure 5a,b and A
O and A
U in
Figure 2b, are responses to the cathodic current waves, C
U in
Figure 5a,b and C
O in
Figure 2b, and can be ascribed to the partial stripping of the electrodeposited Al and Al-V alloys, having naturally different dissolution potentials. This is more apparent in the case of the voltammogram in
Figure 2b, where the peaks A
O probably reflect the dissolution of overpotentially deposited aluminium and some of the Al-V alloys formed concurrently and A
U, which should reflect the dissolution of the Al-V alloy made during Al UPD onto vanadium working electrode,
Figure 2b, and on the vanadium previously deposited onto GC working electrode,
Figure 5a,b. The potentials of those peaks (A
O and A
U) are close to the equilibrium potentials (recorded by potentiodynamic polarization curve in
Figure 2a marked as C
O at 0.000 V and 0.080 V and C
U ≈ 0.350 V vs. Al, respectively.
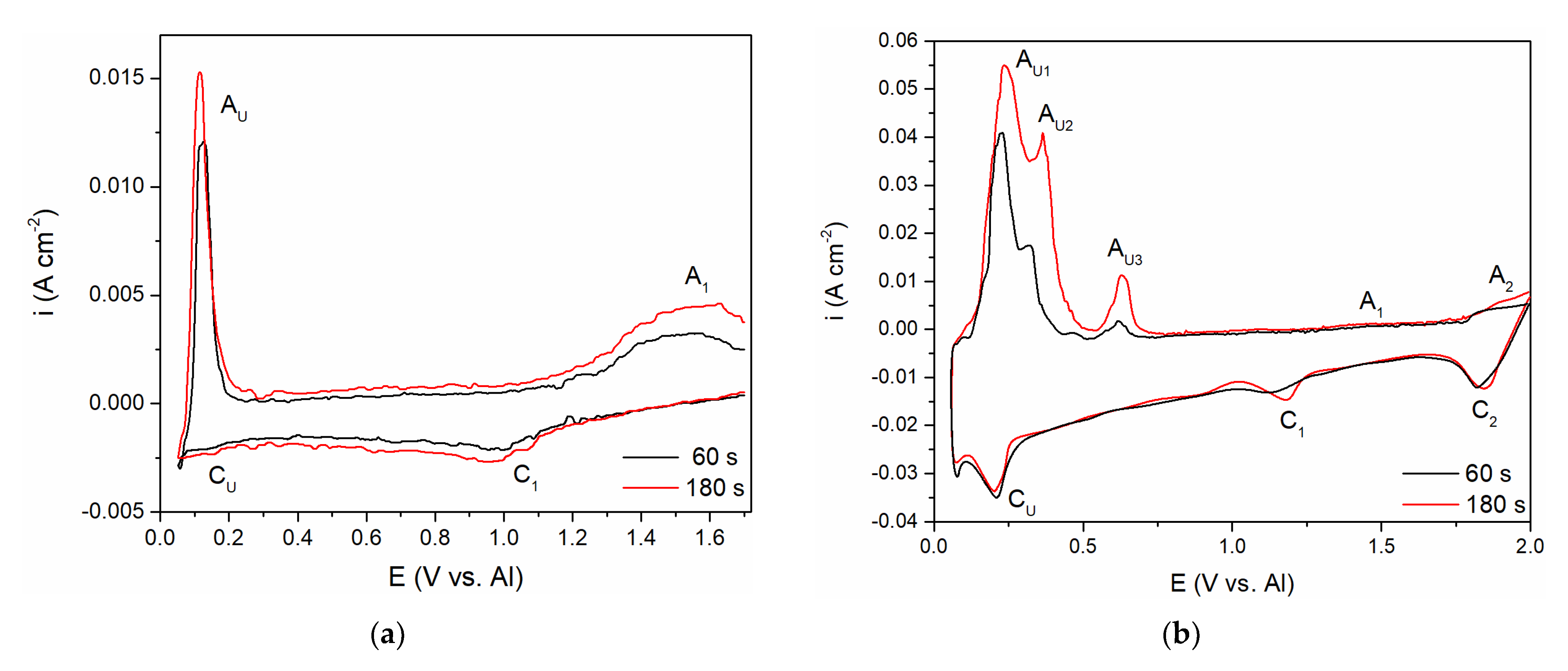
To follow up on the aluminium and vanadium deposition/dissolution processes noticed with CV on GC electrode in the equimolar AlCl
3 + NaCl melt containing V ions added to the electrolyte either by vanadium anodic dissolution or by VCl
3 addition, the voltammetry applying different holding times at chosen cathodic end potentials, E
CF, before returning to the starting potential was recorded,
Figure 6a,b. In both cases, the cathodic end potential was held at 0.050 V vs. Al. In these sets of experiments, where the cathodic end potential E
CF was not negative to the aluminium equilibrium potential, the anodic current waves reflecting the dissolution processes appeared at the potentials very similar to the values recorded on CVs presented in
Figure 2b and
Figure 5. However, under the same experimental conditions, the cathodic and anodic peaks of the voltammograms recorded in the equimolar melt with VCl
3 added, were more clearly defined (A
U1, A
U2, and A
U3), and maxima of the anodic and cathodic peak currents were higher. Generally, the increased deposition time generated an increase in the maximum peak current and charge encompassed by the anodic current peaks. This suggests that with prolonged deposition time under the constant potential applied, quantity of vanadium and aluminium electrodeposited increases, and that Al-V intermetallics are formed between vanadium and underpotentially deposited aluminium. Actually, aluminium is deposited by underpotential deposition on the vanadium that was previously deposited in the quantity which dominates the GC surface [
8].
Aluminium underpotential deposition with previously deposited vanadium enters into solid-state interdiffusion processes and first quantities of Al-V intermetallics are synthetized. Prolonged vanadium deposition and aluminium underpotential deposition provide additional material for more alloy formation. Obviously, there was not only one intermetallic synthetized, because there were several anodic dissolution peaks (A
U1, A
U2, A
U3) reflecting different intermetallics having different dissolution potentials [
8,
9].
3.3. Characterization of Electrodeposited Al‒V Intermetallic
Surface morphologies of the Al-V alloys produced by potentiostatic aluminium underpotential and vanadium codeposition from equimolar AlCl
3 + NaCl melt containing anodically dissolved vanadium at 0.020 V vs. Al, and from the equimolar AlCl
3 + NaCl melt with vanadium added as VCl
3 at 0.050 V vs. Al, are presented
Figure 7 and
Figure 8, respectively.
Presence of the aluminium and vanadium in the deposits were reported by EDS semi-quantitative analysis in the samples presented in
Figure 7 and
Figure 8. The deposit produced by potentiostatic electrodeposition from electrolyte containing anodically dissolved vanadium, besides oxygen, which was impossible to avoid during sample handling, has shown the content of aluminium at 38.55 at. % and vanadium of 23.15 at. %. Content of aluminium was 66.29 at. % and of vanadium 29.18 at. % in the sample obtained by electrodeposition from the electrolyte with vanadium added by VCl
3. According to the EDS analysis, the deposits were free from chloride contamination. EDS data presented were collected from the entire surface given in
Figure 7a and
Figure 8a.
SEM photographs in
Figure 7a–c represent a deposit obtained on GC electrode by potentiostatic aluminium underpotential deposition at 0.020 V vs. Al from the electrolyte containing anodically dissolved vanadium. The micrographs expose a relatively smooth and compact surface coating, well adhered to the working substrate. However, a deposit obtained on GC electrode by the deposition from the electrolyte with vanadium added by VCl
3 at 0.050 V vs. Al, exhibited almost uniformly distributed 3D forms of the cauliflower-like structures,
Figure 8a–c. Individual forms seem to be constructed from approximately spherical grains as the basic element,
Figure 8c. From
Figure 8a–c, it appears that they have been closely packed, with the average particles size around 5 µm, and completely covered the GC surface area.
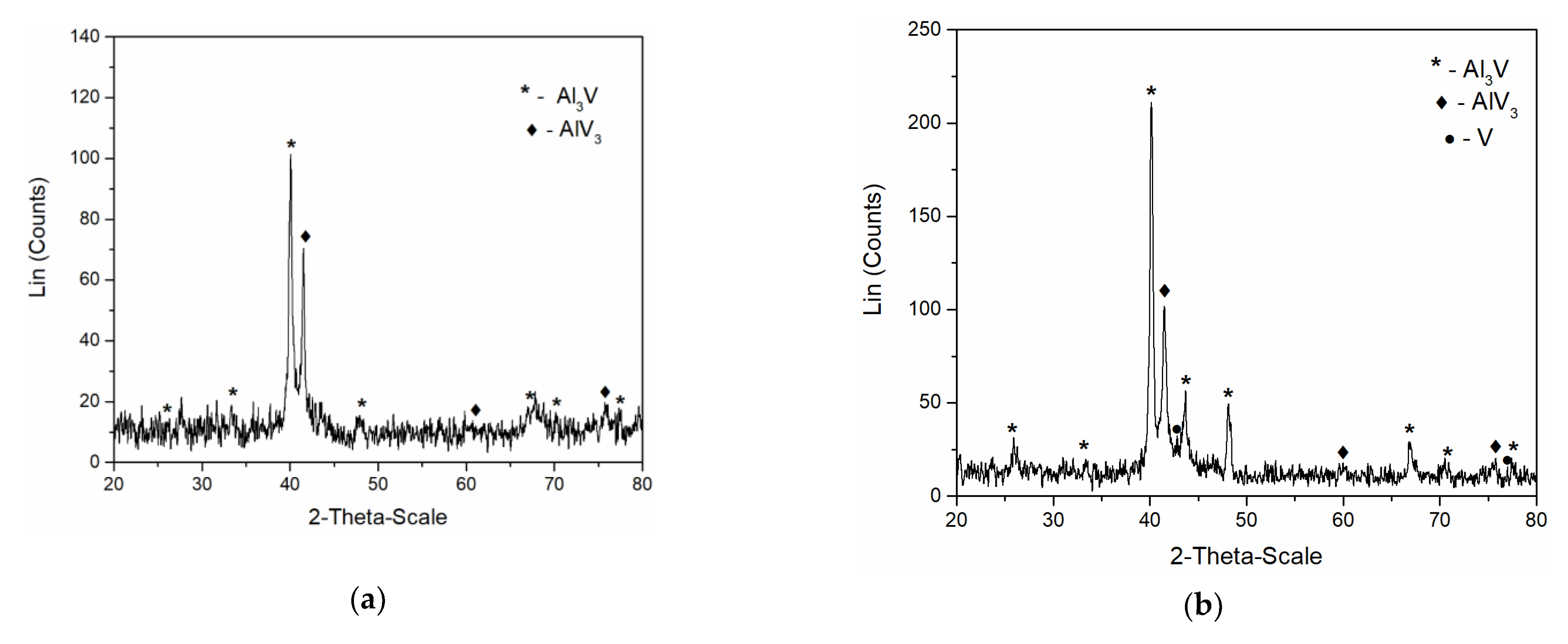
In order to identify the intermetallics synthetized during simultaneous vanadium deposition and aluminium underpotential deposition, XRD analysis of the deposit obtained on the GC working electrode at the potential of 0.020 V vs. Al applied for 5 h in the electrolyte containing anodically dissolved V was conducted. The analysis identified Al
3V and AlV
3 as present in the sample,
Figure 9a. Peaks identified in the diffractogram at 2θ = 25.87°, 33.50°, 40.07°, 48.11°, 66.83°, 70.41°, and 77.47° can be indexed to (101), (110), (112), (200), (204), (220), and (116) reflections from body-centered tetragonal Al
3V [JCPDS No. 03-065-2664]. The diffraction peaks at 2θ = 41.73°, 60.50°, 76.19° are indexed to (110), (200) and (211) reflections from body-centered cubic AlV
3 [JCPDS No. 03-065-5142].
Very similar to these results were the XRD recordings of the deposit obtained on the GC working electrode at constant potential of 0.050 V vs. Al for 5 h, in the electrolyte where as a source of V ions, VCl
3 was used,
Figure 9b. Apart from the prominent peaks corresponding to Al
3Vand AlV
3 alloys, the XRD data indicated the traces of metallic V (2θ peaks at 42.28° and 77.20° [JCPDS No. 01-089-3059]) in the deposit.
Although the Al‒V intermetallics are well characterized, the Al‒V binary phase diagram still contains many uncertainties [
7,
8,
12]. On the basis of binary Al‒V phase diagram, four intermetallics can be identified among Al-rich alloys: Al
21V
2, A1
45V
7, A1
23V
4, and Al
3V, none of which can be formed at the temperatures below 600 °C. Al
3V, an aluminium rich compound, should occur in Al‒V alloys containing 1–25 wt.% V with D0
22 structure prepared at temperature above 746 °C [
12].
Literature reports on metallographic and microprobe analyses made on dilute alloys heat treatment for short times at temperatures between 500 °C and 740 °C, determined that Al
3V phase is an equilibrium phase with a tetragonal structure. Recently, it was reported that the alloy Al
3V was synthesized at temperatures from 1000 °C to 1400 °C from Al and relatively high vanadium content of 3–4wt.% [
13,
26]. At temperature from 1000 °C to 1100 °C and all the way up to 1400 °C the amount of Al
3V increased.
As stated in a recent study [
13], vanadium content in the melt was an important factor influencing the microstructure and controlling the types of V-containing phases formed in Al‒V alloys during rapid solidification process. Al
3V alloy was not formed until vanadium content reached at least 3 wt.% in the Al-V alloy [
13,
27]. The dendritic Al
3V alloy was formed at higher vanadium content in the system, with the boundaries of the Al
3V phase spherical in shape.
In other words, alloys Al3V and AlV3 recorded in the analysis made on the deposit formed by the electrodeposition of vanadium and aluminium from the equimolar AlCl3 + NaCl melt containing vanadium ions onto GC, at applied temperature (200 °C) should not have been synthesized. However, it appears that in this case, potentiostatic electrodeposition is responsible for the controllable synthesis of the alloys. In our study, the deposits produced under all conditions being the same except the vanadium ion concentration in the electrolyte were identified as Al3V and AlV3. It appears that the vanadium concentrations in the electrolyte used were adequate for Al3V and AlV3 alloy formation. However, from the melt containing higher vanadium concentration in the electrolytes used, the deposit displayed a dendritic structure.
Our experiments have confirmed Al-V alloy generation by controlled aluminium and vanadium codeposition in the aluminium underpotential as well as overpotential region of the working potentials. The Al and V codeposition in the region of aluminium overpotentials will be a subject of another report. Nevertheless, it should be noted that the limiting factor in the Al and V codeposition in both aluminium overpotential and aluminium underpotential regions, is the vanadium ion concentration in the electrolyte used. The vanadium ion concentration in the electrolyte used dictates the largest vanadium deposition rate i.e., the mass transfer-controlled vanadium deposition current density. In the same time the vanadium dissolution in the chloride molten salts media is restricted to rather small values [
15,
16,
25,
28].

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








