

Environmental Adaptation of Genetically Uniform Organisms with the Help of Epigenetic Mechanisms—An Insightful Perspective on Ecoepigenetics


Abstract
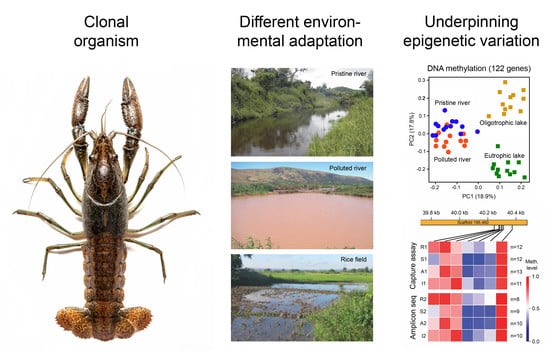
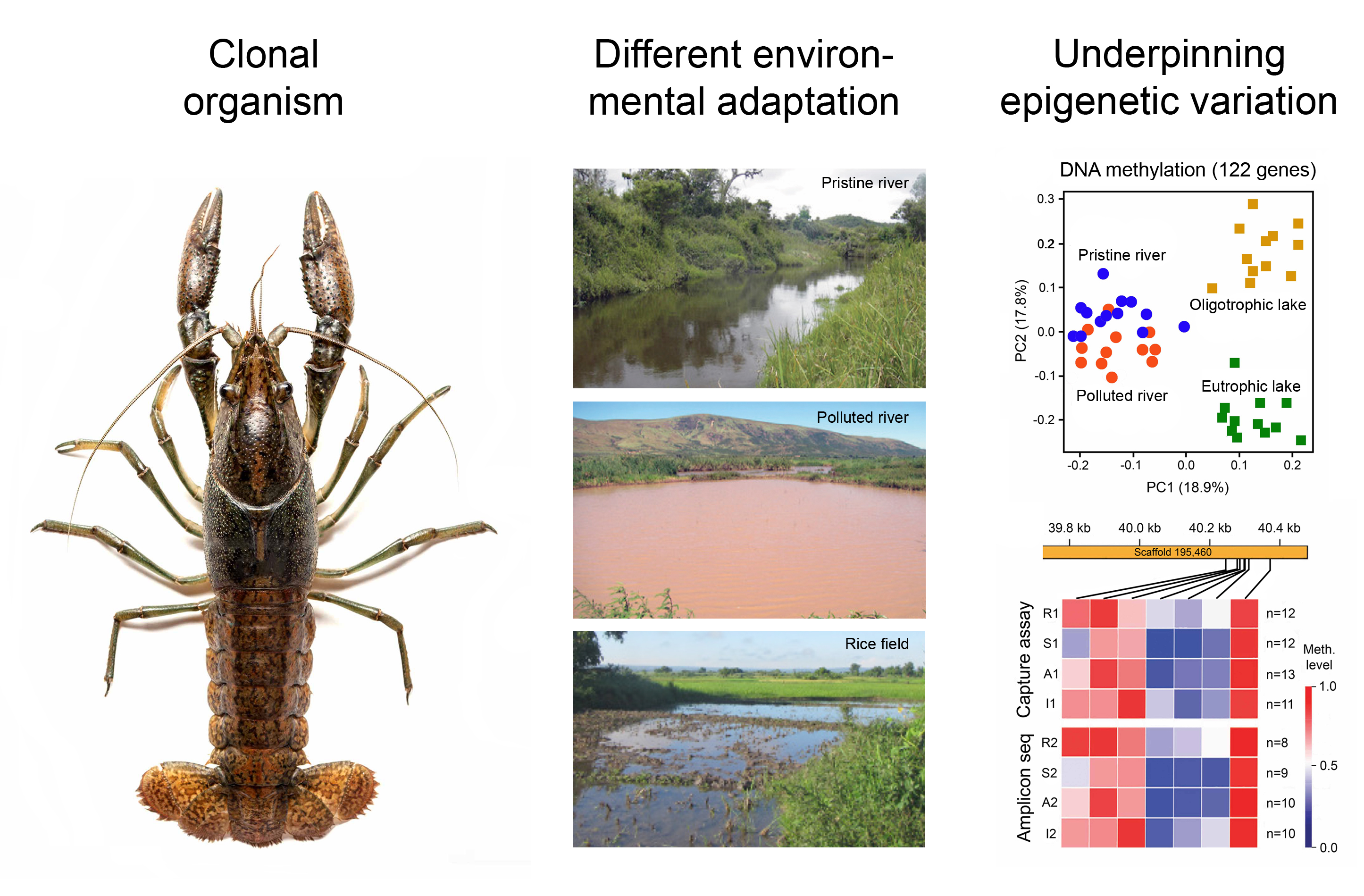
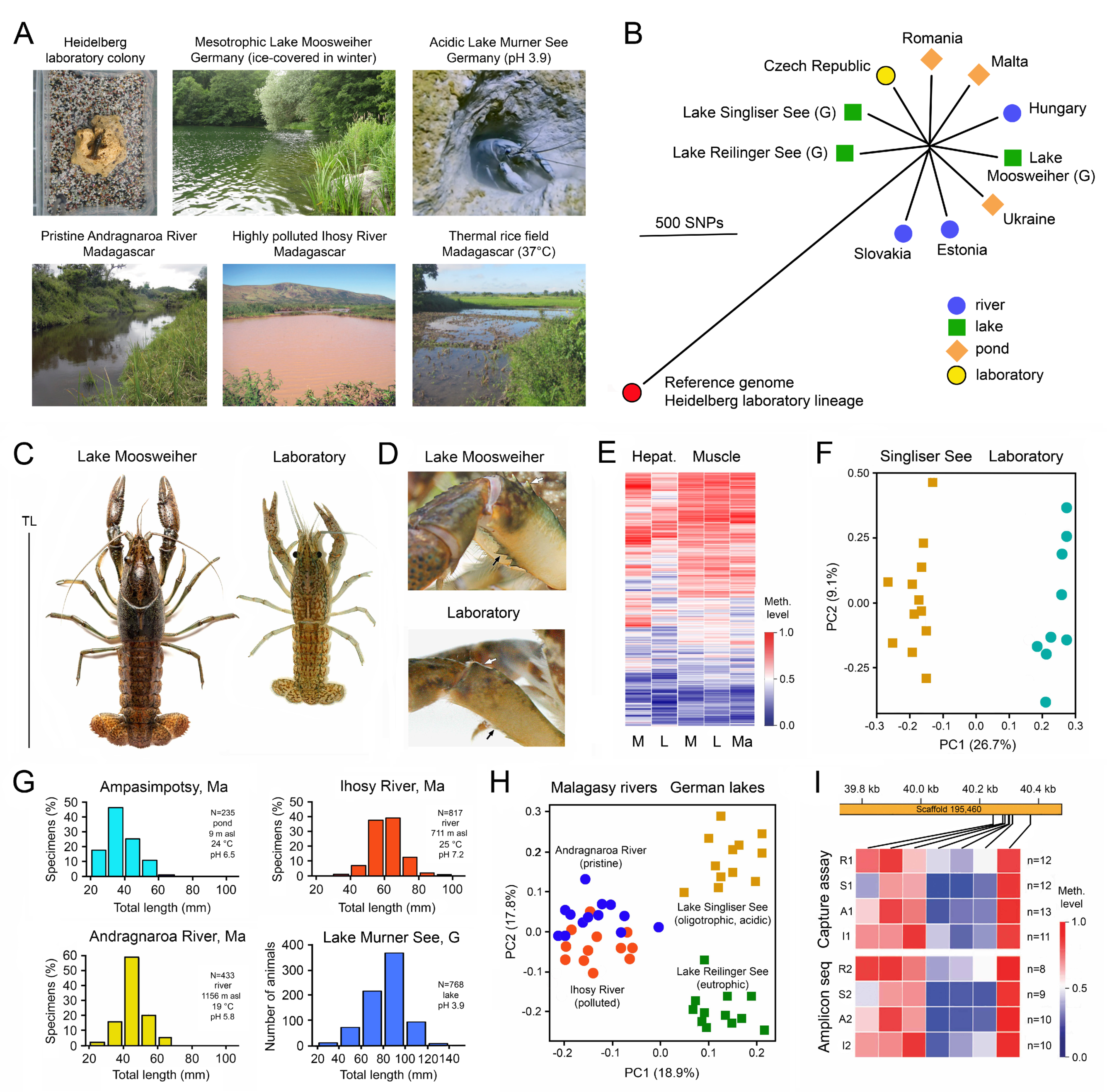
1. Introduction
2. Generation of Phenotypic Diversity in Populations
2.1. Generation of Phenotypic Variation by Genetic Mechanisms
2.2. Generation of Phenotypic Variation by Epigenetic Mechanisms
2.3. Stochastic and Environmentally-Induced Epimutations and Related Phenotypic Change
3. Environmental Adaptation of Clonal Organisms with the Help of Epigenetic Mechanisms
3.1. Case Studies with Animals

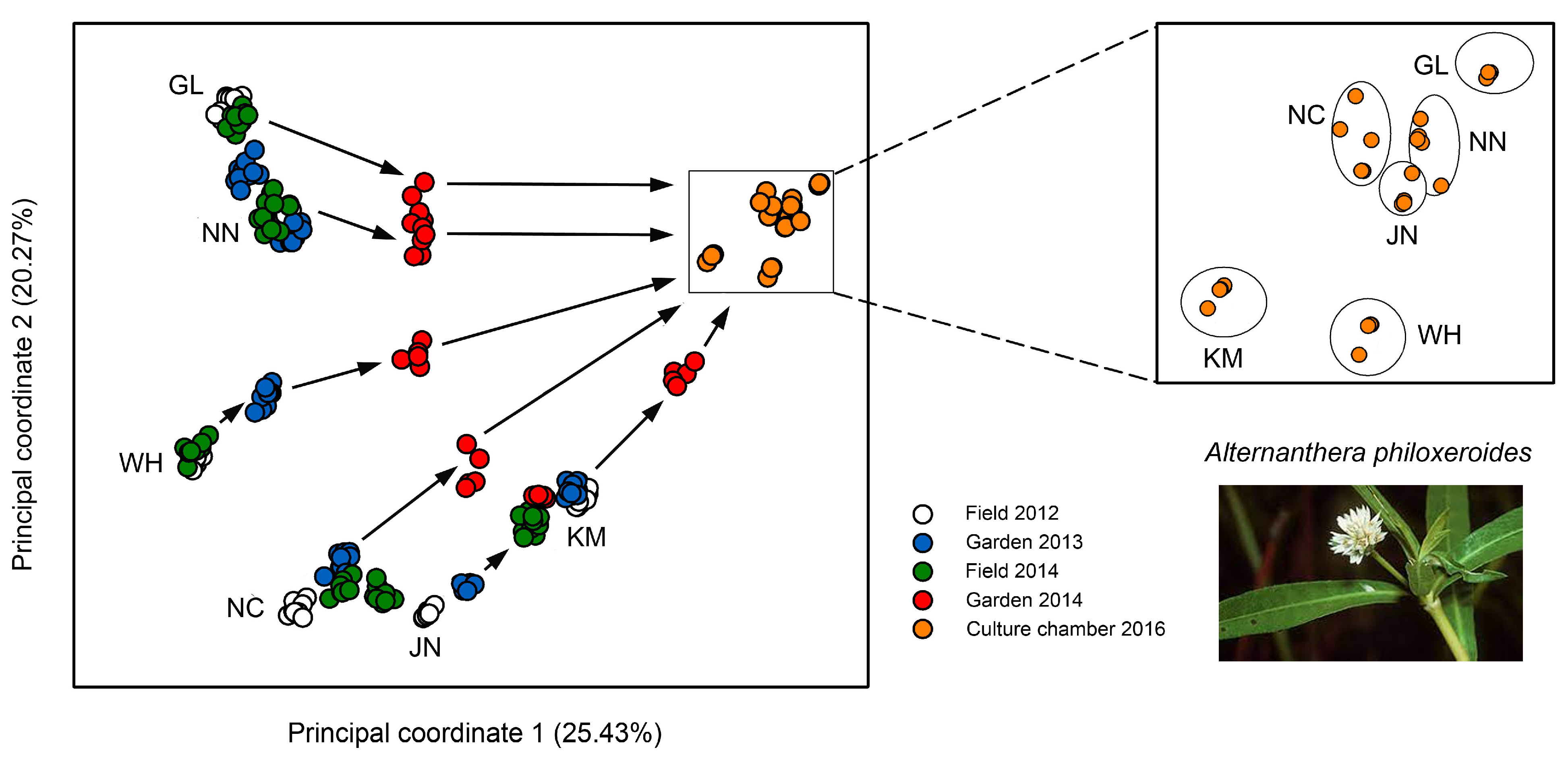
3.2. Case Studies with Plants
3.3. Case Studies with Fungi, Protists and Bacteria
4. Environmental Adaptation of Genetically Impoverished Invaders with the Help of Epigenetic Mechanisms
5. Ecological Implications of Epigenetic Diversity for Genetically Uniform Organisms and Possible Evolutionary Consequences
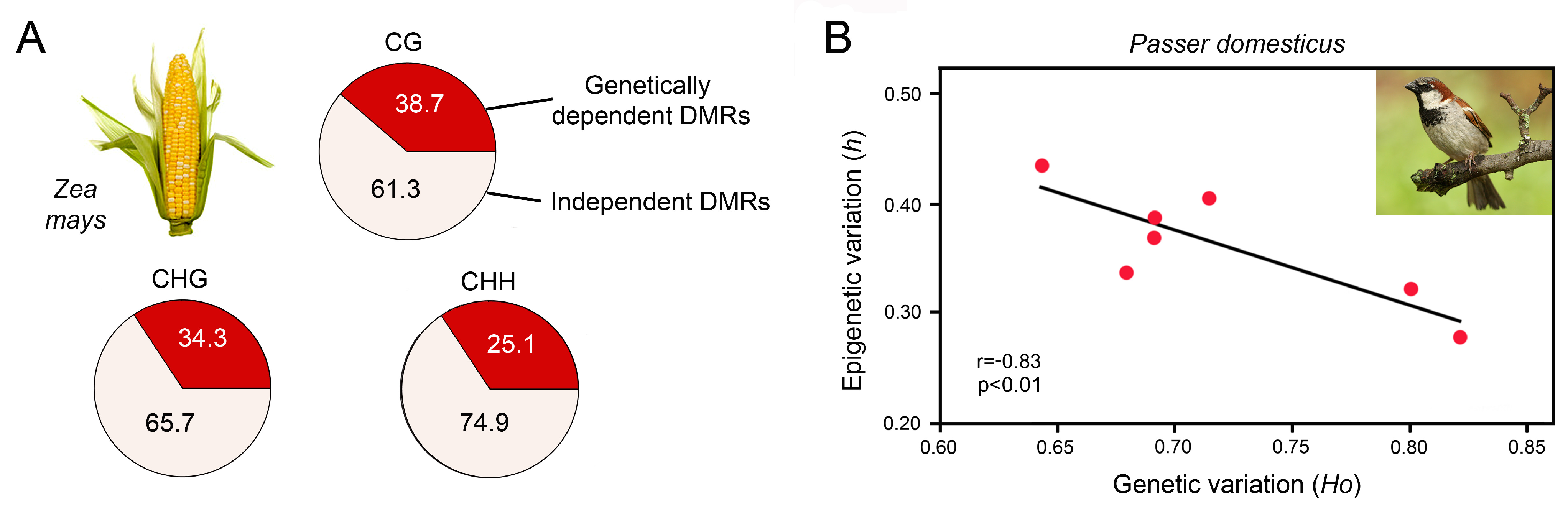
5.1. Capability of Epigenetic Mechanisms to Produce Phenotypic Variation for Environmental Adaptation
5.2. Habitat-Specific Epigenetic Signatures and the Existence of Epigenetic Ecotypes
5.3. Epigenetic Variation as an Explanation of the General-Purpose Genotype and Invasion Paradox
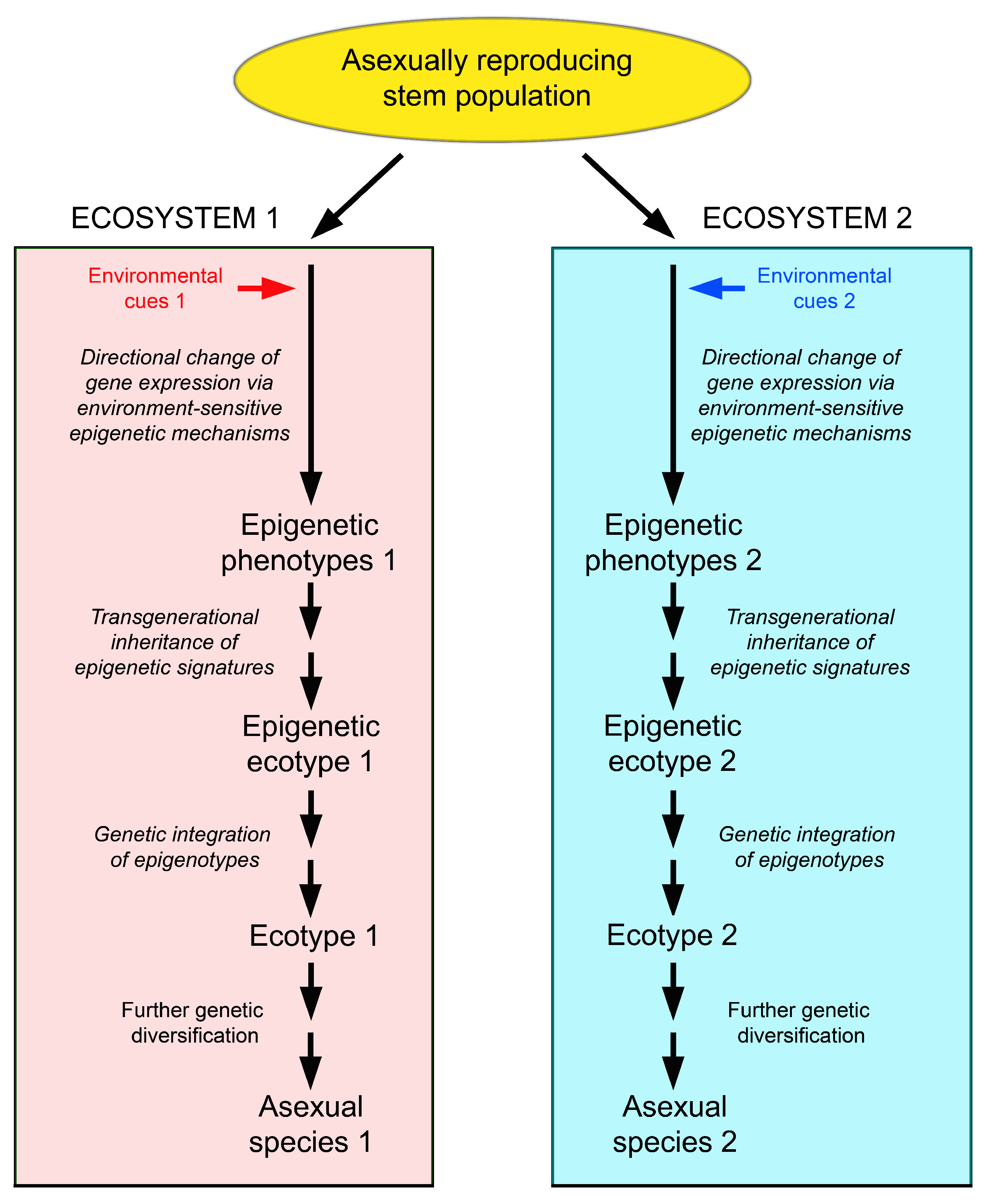
5.4. Relevance of the Production of Epigenetic Variation for the Ecology of Asexually Reproducing Organisms
5.5. Evolutionary Potential of Epigenetically-Based Phenotypes and Epigenetic Ecotypes in Clonal Organisms
6. Short Digression into the Implications of Epigenetic Variation for Environmental Adaptation in Genetically Diverse Animals, Plants and Microorganisms
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeWitt, T.; Scheiner, S.M. (Eds.) Phenotypic Plasticity: Functional and Conceptual Approaches; Oxford University Press: New York, NY, USA, 2004; ISBN 978-0195138962. [Google Scholar]
- Fusco, G.; Minelli, A. Phenotypic plasticity in development and evolution: Facts and concepts. Philos. Trans. R. Soc. Lond. B 2010, 365, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Sommer, R.J. Phenotypic plasticity: From theory and genetics to current and future challenges. Genetics 2020, 215, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, D.W. (Ed.) Phenotypic Plasticity and Evolution. Causes, Consequences, Controversies; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar] [CrossRef]
- Kilvitis, H.J.; Hanson, H.; Schrey, A.W.; Martin, L.B. Epigenetic potential as a mechanism of phenotypic plasticity in vertebrate range expansions. Integr. Comp. Biol. 2017, 57, 385–395. [Google Scholar] [CrossRef]
- Rai, L.S.; Singha, R.; Brahma, P.; Sanyal, K. Epigenetic determinants of phenotypic plasticity in Candida albicans. Fungal Biol. Rev. 2018, 32, 10–19. [Google Scholar] [CrossRef]
- Chapelle, V.; Silvestre, F. Population epigenetics: The extent of DNA methylation variation in wild animal populations. Epigenomes 2022, 6, 31. [Google Scholar] [CrossRef]
- Dar, F.A.; Mushtaq, N.U.; Saleem, S.; Rehman, R.U.; Dar, T.U.; Hakeem, K. Role of epigenetics in modulating phenotypic plasticity against abiotic stresses in plants. Int. J. Genomics 2022, 1092894. [Google Scholar] [CrossRef]
- Vogt, G. Epigenetics and Phenotypic Plasticity in Animals. In Epigenetics, Development, Ecology and Evolution; Vaschetto, L.M., Ed.; Springer: Cham, Switzerland, 2022; pp. 35–108. [Google Scholar] [CrossRef]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. Lond. B 2009, 364, 3403–3418. [Google Scholar] [CrossRef]
- Sahu, P.P.; Pandey, G.; Sharma, N.; Puranik, S.; Muthamilarasan, M.; Prasad, M. Epigenetic mechanisms of plant stress responses and adaptation. Plant Cell Rep. 2013, 32, 1151–1159. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef]
- Duncan, E.J.; Cunningham, C.B.; Dearden, P.K. Phenotypic plasticity: What has DNA methylation got to do with it? Insects 2022, 13, 110. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Kernaleguen, M.; Daviaud, C.; Shen, Y.; Bonnet, E.; Renault, V.; Deleuze, J.-F.; Mauger, F.; Tost, J. Whole-genome bisulfite sequencing for the analysis of genome-wide DNA methylation and hydroxymethylation patterns at single-nucleotide resolution. Meth. Mol. Biol. 2018, 1767, 311–349. [Google Scholar] [CrossRef]
- Peck, J.R.; Yearsley, J.M.; Waxman, D. Explaining the geographic distributions of sexual and asexual populations. Nature 1998, 391, 889–892. [Google Scholar] [CrossRef]
- Tilquin, A.; Kokko, H. What does the geography of parthenogenesis teach us about sex? Philos. Trans. R. Soc. Lond. B 2016, 371, 20150538. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.K. Asexual parasites and their extraordinary host ranges. Integr. Comp. Biol. 2019, 59, 1463–1484. [Google Scholar] [CrossRef]
- Baker, H.G. Characteristics and Modes of Origins of Weeds. In The Genetics of Colonizing Species; Baker, H.G., Stebbins, G.L., Eds.; Academic Press: New York, NY, USA, 1965; pp. 147–172. ISBN 978-0120751501. [Google Scholar]
- Vrijenhoek, R.C. Animal clones and diversity: Are natural clones generalists or specialists? BioScience 1998, 48, 617–628. [Google Scholar] [CrossRef]
- Massicotte, R.; Angers, B. General-purpose genotype or how epigenetics extend the flexibility of a genotype. Genet. Res. Int. 2012, 2012, 317175. [Google Scholar] [CrossRef]
- Sax, D.F.; Brown, J.H. The paradox of invasion. Glob. Ecol. Biogeogr. 2000, 9, 363–371. [Google Scholar] [CrossRef]
- Estoup, A.; Ravigné, V.J.; Hufbauer, R.; Vitalis, R.; Gautier, M.; Facon, B. Is there a genetic paradox of biological invasion? Annu. Rev. Ecol. Evol. Syst. 2016, 47, 51–72. [Google Scholar] [CrossRef]
- Kumarathunge, D.P.; Medlyn, B.E.; Drake, J.E.; Tjoelker, M.G.; Aspinwall, M.J.; Battaglia, M.; Cano, F.C.; Carter, K.R.; Cavaleri, M.A.; Cernusak, L.A.; et al. Acclimation and adaptation components of the temperature dependence of plant photosynthesis at the global scale. New Phytol. 2019, 222, 768–784. [Google Scholar] [CrossRef]
- Ghalambor, C.K.; McKay, J.K.; Carroll, S.P.; Reznick, D.N. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Funct. Ecol. 2007, 21, 394–407. [Google Scholar] [CrossRef]
- Rengefors, K.; Kremp, A.; Reusch, T.B.H.; Wood, A.W. Genetic diversity and evolution in eukaryotic phytoplankton: Revelations from population genetic studies. J. Plankton Res. 2017, 39, 165–179. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.V.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Recombination: The good, the bad and the variable. Philos. Trans. R. Soc. Lond. B 2017, 372, 20170279. [Google Scholar] [CrossRef] [PubMed]
- Low, K.B.; Porter, D.D. Modes of gene transfer and recombination in bacteria. Annu. Rev. Genet. 1978, 12, 249–287. [Google Scholar] [CrossRef] [PubMed]
- Seymour, M.; Räsänen, K.; Kristjánsson, B.K. Drift versus selection as drivers of phenotypic divergence at small spatial scales: The case of Belgjarskógur threespine stickleback. Ecol. Evol. 2019, 9, 8133–8145. [Google Scholar] [CrossRef]
- Clegg, S.M.; Phillimore, A.B. The influence of gene flow and drift on genetic and phenotypic divergence in two species of Zosterops in Vanuatu. Philos. Trans. R. Soc. Lond. B 2010, 365, 1077–1092. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Preite, V. Epigenetic variation in asexually reproducing organisms. Evolution 2014, 68, 644–655. [Google Scholar] [CrossRef]
- Vogt, G. Epigenetic variation in animal populations: Sources, extent, phenotypic implications, and ecological and evolutionary relevance. J. Biosci. 2021, 46, 24. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Frias-Laserre, D.; Villagra, C.A. The importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G. Evolution, Functions and Dynamics of Epigenetic Mechanisms in Animals. In Handbook of Epigenetics: The New Molecular and Medical Genetics, 3rd ed.; Tollefsbol, T., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 521–549. [Google Scholar] [CrossRef]
- Maeji, H.; Nishimura, T. Epigenetic mechanisms in plants. Adv. Bot. Res. 2018, 88, 21–47. [Google Scholar] [CrossRef]
- Madhani, H.D. Unbelievable but true: Epigenetics and chromatin in fungi. Trends Genet. 2021, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, M.A.; Casadesús, J. The bacterial epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20. [Google Scholar] [CrossRef]
- Gentilini, D.; Garagnani, P.; Pisoni, S.; Bacalini, M.G.; Calzari, L.; Mari, D.; Vitale, G.; Franceschi, C.; Di Blasio, A.M. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging 2015, 7, 568–576. [Google Scholar] [CrossRef]
- Plotnikova, O.; Baranova, A.; Skoblov, M. Comprehensive analysis of human microRNA–mRNA interactome. Front. Genet. 2019, 10, 933. [Google Scholar] [CrossRef]
- Nasrullah, A.H.; Ahmed, S.; Rasool, M.; Shah, A.J. DNA methylation across the tree of life, from micro to macro-organism. Bioengineered 2022, 13, 1666–1685. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Kreplova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Gatzmann, F.; Falckenhayn, C.; Gutekunst, J.; Hanna, K.; Raddatz, G.; Coutinho Carneiro, V.; Lyko, F. The methylome of the marbled crayfish links gene body methylation to stable expression of poorly accessible genes. Epigenetics Chromatin 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- He, C.; Zhang, Z.; Li, B.; Tian, S. The pattern and function of DNA methylation in fungal plant pathogens. Microorganisms 2020, 8, 227. [Google Scholar] [CrossRef]
- Nai, Y.-S.; Huang, Y.-C.; Yen, M.-R.; Chen, P.-Y. Diversity of fungal DNA methyltransferases and their association with DNA methylation patterns. Front. Microbiol. 2021, 11, 616922. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Cota, I.; Casadesús, J. DNA methylation in bacteria: From the methyl group to the methylome. Curr. Opin. Microbiol. 2015, 25, 9–16. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.-M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2015, 6, a018762. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhan, Z.; Jiang, D. Histone modifications and their regulatory roles in plant development and environmental memory. J. Genet. Genomics 2019, 46, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Brosch, G.; Loidl, P.; Graessle, S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol. Rev. 2008, 32, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef]
- Moutinho, C.; Esteller, M. MicroRNAs and epigenetics. Adv. Cancer Res. 2017, 135, 189–220. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 40. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Senti, K.A.; Brennecke, J. The piRNA pathway: A fly’s perspective on the guardian of the genome. Trends Genet. 2010, 26, 499–509. [Google Scholar] [CrossRef]
- Li, M.-Z.; Xiao, H.-M.; He, K.; Li, F. Progress and prospects of noncoding RNAs in insects. J. Integr. Agric. 2019, 18, 729–747. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, T.; He, W.; Shen, M.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics 2018, 110, 35–42. [Google Scholar] [CrossRef]
- Waititu, J.K.; Zhang, C.; Liu, J.; Wang, H. Plant non-coding RNAs: Origin, biogenesis, mode of action and their roles in abiotic stress. Int. J. Mol. Sci. 2020, 21, 8401. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S. Role of non-coding RNAs in fungal pathogenesis and antifungal drug responses. Curr. Clin. Microbiol. Rep. 2020, 7, 133–141. [Google Scholar] [CrossRef]
- Stav, S.; Atilho, R.M.; Mirihana Arachchilage, G.; Nguyen, G.; Higgs, G.; Breaker, R.R. Genome-wide discovery of structured noncoding RNAs in bacteria. BMC Microbiol. 2019, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Steffen, P.A.; Ringrose, L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 2014, 15, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Ciabrelli, F.; Comoglio, F.; Fellous, S.; Bonev, B.; Ninova, M.; Szabo, Q.; Xuéreb, A.; Klopp, C.; Aravin, A.; Paro, R.; et al. Stable Polycomb-dependent transgenerational inheritance of chromatin states in Drosophila. Nat. Genet. 2017, 49, 876–886. [Google Scholar] [CrossRef]
- Wright, C.J.; Smith, C.W.J.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.-Z.; Jiang, J.; Duan, C.-G. The crosstalk between epigenetic mechanisms and alternative RNA processing regulation. Front. Genet. 2020, 11, 998. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing – immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Irizarry, R.A. Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. S1), 1757–1764. [Google Scholar] [CrossRef]
- Angers, B.; Perez, M.; Menicucci, T.; Leung, C. Sources of epigenetic variation and their applications in natural populations. Evol. Appl. 2020, 13, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G. Disentangling the Environmentally Induced and Stochastic Developmental Components of Phenotypic Variation. In Phenotypic Switching: Implications in Biology and Medicine; Levine, H., Jolly, M.K., Kulkarni, P., Nanjundiah, V., Eds.; Academic Press: San Diego, CA, USA, 2020; pp. 207–251. [Google Scholar] [CrossRef]
- Shah, J.M. Epimutations and mutations, nurturing phenotypic diversity. Genetica 2022, 150, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G. Facilitation of environmental adaptation and evolution by epigenetic phenotype variation: Insights from clonal, invasive, polyploid, and domesticated animals. Environ. Epigenet. 2017, 3, dvx002. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Breton, S.; Angers, B. Facing environmental predictability with different sources of epigenetic variation. Ecol. Evol. 2016, 6, 5234–5245. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [CrossRef]
- Skinner, M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinol. 2014, 398, 4–12. [Google Scholar] [CrossRef]
- Xue, Y.; Acar, M. Mechanisms for the epigenetic inheritance of stress response in single cells. Curr. Genet. 2018, 64, 1221–1228. [Google Scholar] [CrossRef]
- Liu, J.; He, Z. Small DNA methylation, big player in plant abiotic stress responses and memory. Front. Plant Sci. 2020, 11, 595603. [Google Scholar] [CrossRef]
- Foquet, B.; Castellanos, A.A.; Song, H. Comparative analysis of phenotypic plasticity sheds light on the evolution and molecular underpinnings of locust phase polyphenism. Sci. Rep. 2021, 11, 11925. [Google Scholar] [CrossRef]
- Zhu, T.; Brown, A.P.; Ji, H. The emerging role of ten-eleven translocation 1 in epigenetic responses to environmental exposures. Epigenetics Insights 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Voigt, S.; Kost, L. Differences in temperature-sensitive expression of PcG regulated genes among natural populations of Drosophila melanogaster. G3 Gene Genome Genet. 2021, 11, jkab237. [Google Scholar] [CrossRef] [PubMed]
- Danisman, S. TCP transcription factors at the interface between environmental challenges and the plant’s growth responses. Front. Plant Sci. 2016, 7, 1930. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Ma, C.; Chen, L.; Luo, D.; Chen, R.; Liang, F. Mechanistic insights into the interaction between transcription factors and epigenetic modifications and the contribution to the development of obesity. Front. Endocrinol. 2018, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018, 16, 1419. [Google Scholar] [CrossRef]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Kribelbauer, J.F.; Lu, C.-J.; Rohs, R.; Mann, R.S.; Bussemaker, H.J. Toward a mechanistic understanding of DNA methylation readout by transcription factors. J. Mol. Biol. 2020, 432, 1801–1815. [Google Scholar] [CrossRef]
- Liew, Y.J.; Zoccola, D.; Li, Y.; Tambutté, E.; Venn, A.A.; Michell, C.T.; Cui, G.; Deutekom, E.S.; Kaandorp, J.A.; Voolstra, C.R.; et al. Epigenome-associated phenotypic acclimatization to ocean acidification in a reef-building coral. Sci. Adv. 2018, 4, eaar8028. [Google Scholar] [CrossRef]
- Dybdahl, M.F.; Drown, D.M. The absence of genotypic diversity in a successful parthenogenetic invader. Biol. Invasions 2011, 13, 1663–1672. [Google Scholar] [CrossRef]
- Thorson, J.L.M.; Smithson, M.; Beck, D.; Sadler-Riggleman, I.; Nilsson, E.; Dybdahl, M.; Skinner, M.K. Epigenetics and adaptive phenotypic variation between habitats in an asexual snail. Sci. Rep. 2017, 7, 14139. [Google Scholar] [CrossRef]
- Thorson, J.L.M.; Smithson, M.; Sadler-Riggleman, I.; Beck, D.; Dybdahl, M.; Skinner, M.K. Regional epigenetic variation in asexual snail populations among urban and rural lakes. Environ. Epigenet. 2019, 5, dvz020. [Google Scholar] [CrossRef]
- Vogt, G. Biology, Ecology, Evolution, Systematics and Utilization of the Parthenogenetic Marbled Crayfish, Procambarus virginalis. In Crayfish: Evolution, Habitat and Conservation Strategies; Ribeiro, F.B., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2020; pp. 137–227. ISBN 978-1536169416. [Google Scholar]
- Vogt, G. Studying phenotypic variation and DNA methylation across development, ecology and evolution in the clonal marbled crayfish: A paradigm for investigating epigenotype-phenotype relationships in macro-invertebrates. Sci. Nat. 2022, 109, 16. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G. Phenotypic plasticity and environmental adaptation in the monoclonal marbled crayfish is associated with very little genetic diversity but pronounced epigenetic diversity. Curr. Zool. 2023, zoac094. [Google Scholar] [CrossRef]
- Hossain, M.S.; Patoka, J.; Kouba, A.; Buřič, M. Clonal crayfish as biological model: A review on marbled crayfish. Biologia 2018, 73, 841–855. [Google Scholar] [CrossRef]
- Maiakovska, O.; Andriantsoa, R.; Tönges, S.; Legrand, C.; Gutekunst, J.; Hanna, K.; Pârvulescu, L.; Novitsky, R.; Weiperth, A.; Sciberras, A.; et al. Genome analysis of the monoclonal marbled crayfish reveals genetic separation over a short evolutionary timescale. Commun. Biol. 2021, 4, 74. [Google Scholar] [CrossRef]
- Gutekunst, J.; Andriantsoa, R.; Falckenhayn, C.; Hanna, K.; Stein, W.; Rasamy, J.; Lyko, F. Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nat. Ecol. Evol. 2018, 2, 567–573. [Google Scholar] [CrossRef]
- Gutekunst, J.; Maiakovska, O.; Hanna, K.; Provataris, P.; Horn, H.; Wolf, S.; Skelton, C.E.; Dorn, N.J.; Lyko, F. Phylogeographic reconstruction of the marbled crayfish origin. Commun. Biol. 2021, 4, 1096. [Google Scholar] [CrossRef]
- Vogt, G.; Lukhaup, C.; Pfeiffer, M.; Dorn, N.J.; Williams, B.W.; Schulz, R.; Schrimpf, A. Morphological and genetic characterization of the marbled crayfish, including a determination key. Zootaxa 2018, 4524, 329–350. [Google Scholar] [CrossRef]
- Tönges, S.; Venkatesh, G.; Andriantsoa, R.; Hanna, K.; Gatzmann, F.; Raddatz, F.; Coutinho Carneiro, V.; Lyko, F. Location-dependent DNA methylation signatures in a clonal invasive crayfish. Front. Cell Dev. Biol. 2021, 9, 794506. [Google Scholar] [CrossRef]
- Vogt, G. Functional cytology of the hepatopancreas of decapod crustaceans. J. Morphol. 2019, 280, 1405–1444. [Google Scholar] [CrossRef]
- Andriantsoa, R.; Tönges, S.; Panteleit, J.; Theissinger, K.; Coutinho Carneiro, V.; Rasamy, J.; Lyko, F. Ecological plasticity and commercial impact of invasive marbled crayfish populations in Madagascar. BMC Ecol. 2019, 19, 8. [Google Scholar] [CrossRef]
- Tönges, S.; Masagounder, K.; Lenich, F.; Gutekunst, J.; Tönges, M.; Lohbeck, J.; Miller, A.K.; Böhl, F.; Lyko, F. Evaluating invasive marbled crayfish as a potential livestock for sustainable aquaculture. Front. Ecol. Evol. 2021, 9, 651981. [Google Scholar] [CrossRef]
- Linzmaier, S.M.; Musseau, C.; Matern, S.; Jeschke, J.M. Trophic ecology of invasive marbled and spiny-cheek crayfish populations. Biol. Invasions 2020, 22, 3339–3356. [Google Scholar] [CrossRef]
- Lipták, B.; Veselý, L.; Ercoli, F.; Bláha, M.; Buřič, M.; Ruokonen, T.J.; Kouba, A. Trophic role of marbled crayfish in a lentic freshwater ecosystem. Aquat. Invasions 2019, 14, 299–309. [Google Scholar] [CrossRef]
- Veselý, L.; Ruokonen, T.J.; Weiperth, A.; Kubec, J.; Szajbert, B.; Guo, W.; Ercoli, F.; Bláha, M.; Buřič, M.; Hämäläinen, H.; et al. Trophic niches of three sympatric invasive crayfish of EU concern. Hydrobiol. 2021, 848, 727–737. [Google Scholar] [CrossRef]
- Angers, B.; Schlosser, I.J. The origin of Phoxinus eos-neogaeus unisexual hybrids. Mol. Ecol. 2007, 16, 4562–4571. [Google Scholar] [CrossRef]
- Samantara, K.; Shiv, A.; Sousa, L.L.; Sandhu, K.S.; Priyadarshini, P.; Mohapatra, S. A comprehensive review on epigenetic mechanisms and application of epigenetic modifications for crop improvement. Environ. Exp. Bot. 2021, 188, 104479. [Google Scholar] [CrossRef]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef]
- Dong, B.-C.; Yu, F.-H.; Roiloa, S.R. Ecoepigenetics in clonal and inbreeding plants: Transgenerational adaptation and environmental variation. Front. Plant. Sci. 2019, 10, 622. [Google Scholar] [CrossRef]
- Shi, W.; Chen, X.; Gao, L.; Xu, C.-Y.; Ou, X.; Bossdorf, O.; Yang, J.; Geng, Y. Transient stability of epigenetic population differentiation in a clonal invader. Front. Plant. Sci. 2019, 9, 1851. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Latzel, V.; Fischer, M.; Bossdorf, O. Understanding the evolutionary potential of epigenetic variation: A comparison of heritable phenotypic variation in epiRILs, RILs, and natural ecotypes of Arabidopsis thaliana. Heredity 2018, 121, 257–265. [Google Scholar] [CrossRef]
- Sammarco, I.; Münzbergová, Z.; Latzel, V. DNA methylation can mediate local adaptation and response to climate change in the clonal plant Fragaria vesca: Evidence from a European-scale reciprocal transplant experiment. Front. Plant. Sci. 2022, 13, 827166. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.A.; Yan, J.; et al. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 2019, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Liebl, A.L.; Schrey, A.W.; Richards, C.L.; Martin, L.B. Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr. Comp. Biol. 2013, 53, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Levine, H.; Jolly, M.K.; Kulkarni, P.; Nanjundiah, V. (Eds.) Phenotypic Switching: Implications in Biology and Medicine; Academic Press: San Diego, CA, USA, 2020; ISBN 978-0128179970. [Google Scholar]
- Ghosh, D.; Veeraraghavan, B.; Elangovan, R.; Vivekanandan, P. Antibiotic resistance and epigenetics: More to it than meets the eye. Antimicrob. Agents Chemother. 2020, 64, e02225-19. [Google Scholar] [CrossRef]
- Stajic, D.; Bank, C.; Gordo, I. Adaptive potential of epigenetic switching during adaptation to fluctuating environments. Genome Biol. Evol. 2022, 14, evac065. [Google Scholar] [CrossRef]
- Kronholm, I.; Johannesson, H.; Ketola, T. Epigenetic control of phenotypic plasticity in the filamentous fungus Neurospora crassa. G3 Genes Genomes Genet. 2016, 6, 4009–4022. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, M.U.; Kalsoom, F.; Khan, M.I.; Gao, S.; Unar, A.; Zubair, M.; Bilal, M. Mechanisms of gene regulation by histone degradation in adaptation of yeast: An overview of recent advances. Arch. Microbiol. 2022, 204, 287. [Google Scholar] [CrossRef]
- Weiner, A.G.M.; Katz, L.A. Epigenetics as driver of adaptation and diversification in microbial eukaryotes. Front. Genet. 2021, 12, 642220. [Google Scholar] [CrossRef]
- Huang, R.; Ding, J.; Gao, K.; Cruz de Carvalho, M.H.; Tirichine, L.; Bowler, C.; Lin, X. A potential role for epigenetic processes in the acclimation response to elevated pCO2 in the model diatom Phaeodactylum tricornutum. Front. Microbiol. 2019, 9, 3342. [Google Scholar] [CrossRef]
- Casadesús, J.; Low, D.A. Programmed heterogeneity: Epigenetic mechanisms in bacteria. J. Biol. Chem. 2013, 288, 13929–13935. [Google Scholar] [CrossRef]
- Riber, L.; Hansen, L.H. Epigenetic memories: The hidden drivers of bacterial persistence? Trends Microbiol. 2021, 29, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, J.S.; Khan, N.A.; Maciver, S.K.; Alharbi, A.M.; Alfahemi, H.; Siddiqui, R. Epigenetic-mediated antimicrobial resistance: Host versus pathogen epigenetic alterations. Antibiotics 2022, 11, 809. [Google Scholar] [CrossRef] [PubMed]
- Miro-Blanch, J.; Yanes, O. Epigenetic regulation at the interplay between gut microbiota and host metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Woo, V.; Alenghat, T. Epigenetic regulation by gut microbiota. Gut Microbes 2022, 14, e2022407. [Google Scholar] [CrossRef]
- Kundu, P.; Torres, E.R.S.; Stagaman, K.; Kasschau, K.; Okhovat, M.; Holden, S.; Ward, S.; Nevonen, K.A.; Davis, B.A.; Saito, T.; et al. Integrated analysis of behavioral, epigenetic, and gut microbiome analyses in AppNL-G-F, AppNL-F, and wild type mice. Sci. Rep. 2021, 11, 4678. [Google Scholar] [CrossRef]
- Schrey, A.W.; Coon, C.A.C.; Grispo, M.T.; Awad, M.; Imboma, T.; McCoy, E.D.; Mushinsky, H.R.; Richards, C.L.; Martin, L.B. Epigenetic variation may compensate for decreased genetic variation with introductions: A case study using house sparrows (Passer domesticus) on two continents. Genetics Res. Int. 2012, 2012, 979751. [Google Scholar] [CrossRef]
- Sheldon, E.L.; Schrey, A.; Andrew, S.C.; Ragsdale, A.; Griffith, S.C. Epigenetic and genetic variation among three separate introductions of the house sparrow (Passer domesticus) into Australia. R. Soc. Opensci. 2018, 5, 172185. [Google Scholar] [CrossRef]
- Hu, J.; Askary, A.M.; Thurman, T.J.; Spiller, D.A.; Palmer, T.M.; Pringle, R.M.; Barrett, R. The epigenetic signature of colonizing new environments in anolis lizards. Mol. Biol. Evol. 2019, 36, 2165–2170. [Google Scholar] [CrossRef]
- Mounger, J.; Boquete, M.T.; Schmid, M.W.; Granado, R.; Robertson, M.H.; Voors, S.A.; Langanke, K.L.; Alvarez, M.; Wagemaker, C.A.M.; Schrey, A.W.; et al. Inheritance of DNA methylation differences in the mangrove Rhizophora mangle. Evol. Dev. 2021, 23, 351–374. [Google Scholar] [CrossRef]
- Mounger, J.; Ainouche, M.L.; Bossdorf, O.; Cavé-Radet, A.; Li, B.; Parepa, M.; Armel, S.; Yang, J.; Richards, C.L. Epigenetics and the success of invasive plants. Philos. Trans. R. Soc. B 2021, 376, 20200117. [Google Scholar] [CrossRef]
- Rajpal, V.R.; Rathore, P.; Mehta, S.; Wadhwa, N.; Yadav, P.; Berry, E.; Goel, S.; Bhat, V.; Raina, S.N. Epigenetic variation: A major player in facilitating plant fitness under changing environmental conditions. Front. Cell Dev. Biol. 2022, 10, 1020958. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, S.; Watson, R.; McQuaid, C.D.; Gouws, G.; Porri, F. Epigenetic variation among natural populations of the South African sandhopper Talorchestia capensis. Evol. Ecol. 2017, 31, 77–91. [Google Scholar] [CrossRef]
- Wilschut, R.A.; Oplaat, C.; Snoek, L.B.; Kirschner, J.; Verhoeven, K.J.F. Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol. Ecol. 2016, 25, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.A.; Lauerman, T.; Crawford, D.; DiMichele, L. Genetic mechanisms for adapting to a changing environment. Annu. Rev. Genet. 1991, 25, 629–659. [Google Scholar] [CrossRef]
- Lande, R.; Shannon, S. The role of genetic variation in adaptation and population persistence in a changing environment. Evolution 1996, 50, 434–437. [Google Scholar] [CrossRef]
- Barrett, R.D.H.; Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 2008, 23, 38–44. [Google Scholar] [CrossRef]
- Bradshaw, A.D. Environment and phenotypic plasticity. Brookhaven Symp. Biol. 1974, 25, 75–94. [Google Scholar]
- Scheiner, S.M. Genetics and evolution of phenotypic plasticity. Annu. Rev. Ecol. Syst. 1993, 24, 35–68. [Google Scholar] [CrossRef]
- Felsenfeld, G. A Brief History of Epigenetics. In Epigenetics, 2nd ed.; Allis, C.D., Caparros, M.-L., Jenuwein, T., Reinberg, D., Lachner, M., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 1–10. [Google Scholar] [CrossRef]
- Peaston, A.E.; Whitelaw, E. Epigenetics and phenotypic variation in mammals. Mamm. Genome 2006, 17, 365–374. [Google Scholar] [CrossRef]
- Champagne, F.A.; Curley, J.P. Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neurosci. Biobehav. Rev. 2009, 33, 593–600. [Google Scholar] [CrossRef]
- McGowan, P.O.; Suderman, M.; Sasaki, A.; Huang, T.C.T.; Hallett, M.; Meaney, M.J.; Szyf, M. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE 2011, 6, e14739. [Google Scholar] [CrossRef] [PubMed]
- Campagna, M.P.; Xavier, A.; Lechner-Scott, J.; Maltby, V.; Scott, R.J.; Butzkueven, H.; Jokubaitis, V.G.; Lea, R.A. Epigenome-wide association studies: Current knowledge, strategies and recommendations. Clin. Epigenetics 2021, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Lei, S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E. The evolutionary implications of epigenetic inheritance. Interface Focus 2017, 7, 20160135. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, D.; Venney, C.J.; Bernatchez, L.; Wellenreuther, M. Epigenetic inheritance and reproductive mode in plants and animals. Trends Ecol. Evol. 2021, 36, 1124–1140. [Google Scholar] [CrossRef]
- Skinner, M.K.; Nilsson, E.E. Role of environmentally induced epigenetic transgenerational inheritance in evolutionary biology: Unified evolution theory. Environ. Epigenet. 2021, 7, 1–12. [Google Scholar] [CrossRef]
- Feiner, N.; Radersma, R.; Vasquez, L.; Ringnér, M.; Nystedt, B.; Raine, A.; Tobi, E.W.; Heijmans, B.T.; Uller, T. Environmentally-induced DNA methylation is inherited across generations in an aquatic keystone species (Daphnia magna). iScience 2022, 25, 104303. [Google Scholar] [CrossRef]
- Burggren, W. Epigenetic inheritance and its role in evolutionary biology: Re-evaluation and new perspectives. Biology 2016, 5, 24. [Google Scholar] [CrossRef]
- Gilbert, J.; Blaser, M.; Caporaso, J.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Vannier, N.; Mony, C.; Bittebière, A.-K.; Vandenkoornhuyse, P. Epigenetic mechanisms and microbiota as a toolbox for plant phenotypic adjustment to environment. Front. Plant Sci. 2015, 6, 1159. [Google Scholar] [CrossRef]
- Voolstra, C.R.; Ziegler, M. Adapting with microbial help: Microbiome flexibility facilitates rapid responses to environmental change. Bioessays 2020, 42, e2000004. [Google Scholar] [CrossRef] [PubMed]
- Ghaderiardakani, F.; Quartino, M.L.; Wichard, T. Microbiome-dependent adaptation of seaweeds under environmental stresses: A perspective. Front. Mar. Sci. 2020, 7, 575228. [Google Scholar] [CrossRef]
- Chen, C.; Wang, M.; Zhu, J.; Tang, Y.; Zhang, H.; Zhao, Q.; Jing, M.; Chen, Y.; Xu, X.; Jiang, J.; et al. Long-term effect of epigenetic modification in plant–microbe interactions: Modification of DNA methylation induced by plant growth-promoting bacteria mediates promotion process. Microbiome 2022, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Van Doninck, K.; Schön, I.; De Bruyn, L.; Martens, K. A general purpose genotype in an ancient asexual. Oecologia 2002, 132, 205–212. [Google Scholar] [CrossRef]
- Drown, D.; Levri, E.P.; Dybdahl, M.F. Invasive genotypes are opportunistic specialists not general purpose genotypes. Evol. Appl. 2011, 4, 132–143. [Google Scholar] [CrossRef]
- Pimentel, D.; McNair, S.; Janecka, J.; Wightman, J.; Simmonds, C.; O’Connell, C.; Wong, E.; Russel, L.; Zern, J.; Aquino, T.; et al. Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ. 2001, 84, 1–20. [Google Scholar] [CrossRef]
- Davis, M.A. Invasion Biology; Oxford University Press: Oxford, UK, 2009; ISBN 978-0199218769. [Google Scholar]
- Hawes, N.A.; Fidler, A.E.; Dunphy, B.J.; Smith, K.F.; Tremblay, L.A.; Pochon, X. Understanding the role of DNA methylation in successful biological invasions: A review. Biol. Invasions 2018, 20, 2285–2300. [Google Scholar] [CrossRef]
- Coutinho Carneiro, V.; Lyko, F. Rapid epigenetic adaptation in animals and its role in invasiveness. Integr. Comp. Biol. 2020, 60, 267–274. [Google Scholar] [CrossRef]
- Suomalainen, E.; Saura, A.; Lokki, J. Cytology and Evolution in Parthenogenesis; CRC Press: Boca Raton, FL, USA, 1987; ISBN 0849359813. [Google Scholar]
- De Meeûs, T.; Prugnolle, F.; Agnew, P. Asexual reproduction: Genetics and evolutionary aspects. Cell. Mol. Life Sci. 2007, 64, 1355–1372. [Google Scholar] [CrossRef]
- Schön, I.; Martens, K.; Dijk, P. (Eds.) Lost sex: The Evolutionary Biology of Parthenogenesis; Springer: Dordrecht, The Netherlands, 2009; ISBN 978-9400779969. [Google Scholar]
- Butlin, R.; Schön, I.; Martens, K. Asexual reproduction in nonmarine ostracods. Heredity 1998, 81, 473–480. [Google Scholar] [CrossRef]
- Eckert, C.G. The loss of sex in clonal plants. Evol. Ecol. 2002, 15, 501–520. [Google Scholar] [CrossRef]
- Taylor, J.W.; Jacobson, D.J.; Fisher, M.C. The evolution of asexual fungi: Reproduction, speciation and classification. Annu. Rev. Phytopathol. 1999, 37, 197–246. [Google Scholar] [CrossRef] [PubMed]
- De Kroon, H.; van Groenendael, J. (Eds.) The Ecology and Evolution of Clonal Plants; Backhuys Press: Leiden, The Netherlands, 1997; ISBN 978-9401713450. [Google Scholar]
- Chang, Z.; Yadav, V.; Lee, S.C.; Heitman, J. Epigenetic mechanisms of drug resistance in fungi. Fungal Genet. Biol. 2019, 132, 103253. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Jeon, J. Epigenetic regulation of development and pathogenesis in fungal plant pathogens. Mol. Plant. Pathol. 2017, 18, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; Vavouri, T. Mechanisms of epigenetic inheritance of variable traits through the germline. Reproduction 2020, 159, R251–R263. [Google Scholar] [CrossRef] [PubMed]
- Harney, E.; Paterson, S.; Collin, H.; HH Chan, B.H.K.; Bennett, D.; Plaistow, S.J. Pollution induces epigenetic effects that are stably transmitted across multiple generations. Evol. Lett. 2022, 6, 118–135. [Google Scholar] [CrossRef]
- Perales, R.; Pagano, D.; Wan, G.; Fields, B.D.; Saltzman, A.L.; Kennedy, S.G. Transgenerational epigenetic inheritance is negatively regulated by the HERI-1 chromodomain protein. Genetics 2018, 210, 1287–1299. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef]
- Kronholm, I.; Bassett, A.; Baulcombe, D.; Collins, S. Epigenetic and genetic contributions to adaptation in Chlamydomonas. Mol. Biol. Evol. 2017, 34, 2285–2306. [Google Scholar] [CrossRef]
- Kronholm, I.; Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 2016, 25, 18561868. [Google Scholar] [CrossRef] [PubMed]
- Liew, Y.J.; Howells, E.J.; Wang, X.; Michell, C.T.; Burt, J.A.; Idaghdour, Y.; Aranda, M. Intergenerational epigenetic inheritance in reef-building corals. Nat. Clim. Chang. 2020, 10, 254–259. [Google Scholar] [CrossRef]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Integration of sperm ncRNA-directed DNA methylation and DNA methylation-directed histone retention in epigenetic transgenerational inheritance. Epigenetics Chromatin 2021, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Houri-Ze’evi, L.; Rechavi, O. A matter of time: Small RNAs regulate the duration of epigenetic inheritance. Trends Genet. 2017, 33, 46–57. [Google Scholar] [CrossRef]
- Waddington, C.H. Genetic assimilation of an acquired character. Evolution 1953, 7, 118–126. [Google Scholar] [CrossRef]
- Ehrenreich, I.M.; Pfennig, D.W. Genetic assimilation: A review of its potential proximate causes and evolutionary consequences. Ann. Bot. 2016, 117, 769–779. [Google Scholar] [CrossRef]
- Hanson, H.E.; Liebl, A.L. The mutagenic consequences of DNA methylation within and across generations. Epigenomes 2022, 6, 33. [Google Scholar] [CrossRef]
- Lutsenko, E.; Bhagwat, A.S. Principal causes of hot spots for cytosine to thymine mutations at sites of cytosine methylation in growing cells. A model, its experimental support and implications. Mut. Res. 1999, 437, 11–20. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Piferrer, F. Epimutations in developmental genes underlie the onset of domestication in farmed European Sea Bass. Mol. Biol. Evol. 2019, 36, 2252–2264. [Google Scholar] [CrossRef]
- Mark Welch, D.B.; Meselson, M. Evidence for the evolution of bdelloid rotifers without sexual reproduction or genetic exchange. Science 2000, 288, 1211–1215. [Google Scholar] [CrossRef]
- Lowry, D.B. Ecotypes and the controversy over stages in the formation of new species. Biol. J. Linn. Soc. 2012, 106, 241–257. [Google Scholar] [CrossRef]
- Childebayeva, A.; Harman, T.; Weinstein, J.; Day, T.; Brutsaert, T.D.; Bigham, A.W. Genome-wide DNA methylation changes associated with high-altitude acclimatization during an Everest base camp trek. Front. Physiol. 2021, 12, 660906. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, Z.-B.; Chen, J.; Huang, X.-F.; Li, X.-M.; Liang, Y.-B.; Mao, J.-Y.; Chen, X.; Zheng, Z.; Bakshi, A.; et al. Genetic signatures of high-altitude adaptation in Tibetans. Proc. Natl. Acad. Sci. USA 2017, 114, 4178–4194. [Google Scholar] [CrossRef]
- Basak, N.; Thangaraj, K. High-altitude adaptation: Role of genetic and epigenetic factors. J. Biosci. 2021, 46, 107. [Google Scholar] [CrossRef] [PubMed]
- Galen, S.C.; Natarajan, C.; Moriyama, H.; Weber, R.E.; Fago, A.; Benham, P.M.; Chavez, A.N.; Cheviron, Z.A.; Storz, J.F.; Witt, C.C. Contribution of a mutational hot spot to hemoglobin adaptation in high-altitude Andean house wrens. Proc. Natl. Acad. Sci. USA 2015, 112, 13958–13963. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, S.; Saldías, G.S.; Vargas, C.A.; Porri, F. Long term environmental variability modulates the epigenetics of maternal traits of kelp crabs in the coast of Chile. Sci. Rep. 2022, 12, 18806. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.; Wilson, D.J.; Feil, E.; Eyre-Walker, A. The distribution of bacterial doubling times in the wild. Proc. R. Soc. B 2016, 285, 20180789. [Google Scholar] [CrossRef]
- Lacey, E.P. Onset of reproduction in plants: Size-versus age-dependency. Trends Ecol. Evol. 1986, 1, 72–75. [Google Scholar] [CrossRef]
- Ebert, D. Daphnia as a versatile model system in ecology and evolution. EvoDevo 2022, 13, 16. [Google Scholar] [CrossRef]
- Matkin, C.O.; Testa, J.W.; Ellis, G.M.; Saulitis, E.L. Life history and population dynamics of southern Alaska resident killer whales (Orcinus orca). Mar. Mamm. Sci. 2014, 30, 460–479. [Google Scholar] [CrossRef]
- Lee, P.C.; Fishlock, V.; Webber, C.E.; Moss, C.J. The reproductive advantages of a long life: Longevity and senescence in wild female African elephants. Behav. Ecol. Sociobiol. 2016, 70, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Puckett, E.E.; Munshi-South, J. Brown rat demography reveals pre-commensal structure in eastern Asia before expansion into Southeast Asia. Genome Res. 2019, 29, 762–770. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Mechanisms (Act by DNA Sequence or Frequency Change) | Epigenetic Mechanisms (Act without DNA Sequence Change) |
|---|---|
| Mutation | DNA methylation |
| Recombination | Histone modifications |
| Genetic drift | Non-coding RNAs |
| Gene flow | Polycomb/Trithorax system |
| Alternative splicing 1 | mRNA editing |
| mRNA modifications |
| Species | Epigenetic Mechanism | Reference |
|---|---|---|
| Animals | ||
| Stylophora pistillataa | DNA methylation | Liew et al. [93] |
| Procambarus virginalisa | DNA methylation | Tönges et al. [105] |
| Potamopyrgus antipodaruma | DNA methylation | Thorson et al. [95] |
| Potamopyrgus antipodaruma | DNA methylation | Thorson et al. [96] |
| Chrosomus eos-neogaeusa | DNA methylation | Massicotte and Angers [21] |
| Chrosomus eos-neogaeusa | DNA methylation | Leung et al. [80] |
| Anolis sagreib | DNA methylation | Hu et al. [136] |
| Passer domesticusb | DNA methylation | Schrey et al. [134] |
| Passer domesticusb | DNA methylation | Liebl et al. [120] |
| Plants | ||
| Alternanthera philoxeroidesa | DNA methylation | Shi et al. [116] |
| Fragaria vescaa | DNA methylation | Sammarco et al. [118] |
| Taraxacum officinalea | DNA methylation | Wilschut et al. [141] |
| Arabidopsis thalianac | DNA methylation | Zhang et al. [117] |
| Zea maysc | DNA methylation | Xu et al. [119] |
| Rhizophora mangleb | DNA methylation | Mounger et al. [137] |
| Various speciesb | Various mechanisms | Mounger et al. [138] |
| Various speciesb | Various mechanisms | Rajpal et al. [139] |
| Fungi | ||
| Neurospora crassaa | Histone modifications | Kronholm et al. [124] |
| Candida albicansa | Histone modifications | Rai et al. [6] |
| Saccharomyces spec.a | Histone modifications | Khan et al. [125] |
| Protists | ||
| Various speciesa | Various mechanisms | Weiner and Katz [126] |
| Phaeodactylum tricornutuma | Histone modif., ncRNAs | Huang et al. [127] |
| Bacteria | ||
| Various speciesa | DNA methylation | Casadesús and Low [128] |
| Escherichia colia | DNA methylation | Ghosh et al. [122] |
| Escherichia colia | DNA methylation | Riber and Hansen [129] |
| Various speciesa | DNA methylation | Muhammad et al. [130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, G. Environmental Adaptation of Genetically Uniform Organisms with the Help of Epigenetic Mechanisms—An Insightful Perspective on Ecoepigenetics. Epigenomes 2023, 7, 1. https://doi.org/10.3390/epigenomes7010001
Vogt G. Environmental Adaptation of Genetically Uniform Organisms with the Help of Epigenetic Mechanisms—An Insightful Perspective on Ecoepigenetics. Epigenomes. 2023; 7(1):1. https://doi.org/10.3390/epigenomes7010001
Chicago/Turabian StyleVogt, Günter. 2023. "Environmental Adaptation of Genetically Uniform Organisms with the Help of Epigenetic Mechanisms—An Insightful Perspective on Ecoepigenetics" Epigenomes 7, no. 1: 1. https://doi.org/10.3390/epigenomes7010001
APA StyleVogt, G. (2023). Environmental Adaptation of Genetically Uniform Organisms with the Help of Epigenetic Mechanisms—An Insightful Perspective on Ecoepigenetics. Epigenomes, 7(1), 1. https://doi.org/10.3390/epigenomes7010001