
One Omics Approach Does Not Rule Them All: The Metabolome and the Epigenome Join Forces in Haematological Malignancies

 , , ,
, , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Different Levels of Epigenetic Regulation with the Ability to Orchestrate Gene Expression
2. Epigenetic Alterations in Haematopoietic Tumours
2.1. DNA Methylation Dysregulation in Haematological Malignancies
2.2. Dysregulations of Histone Modifiers in Haematological Malignancies
2.3. The Roles of miRNAs in Haematological Malignancies
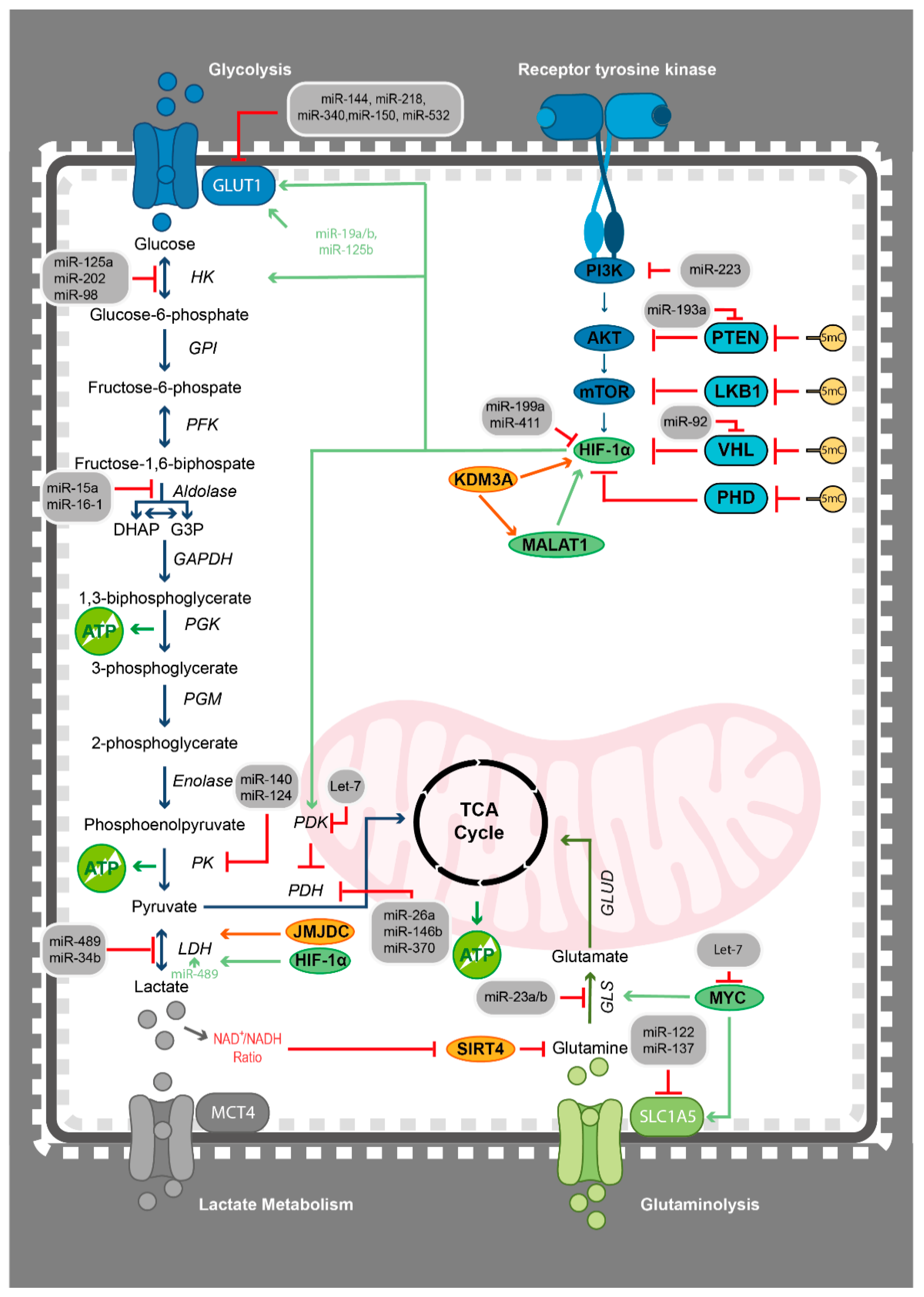
3. The Cellular Metabolism in Haematological Malignancies
4. The Teamwork between Epigenetics and Metabolism in Haematological Malignancies
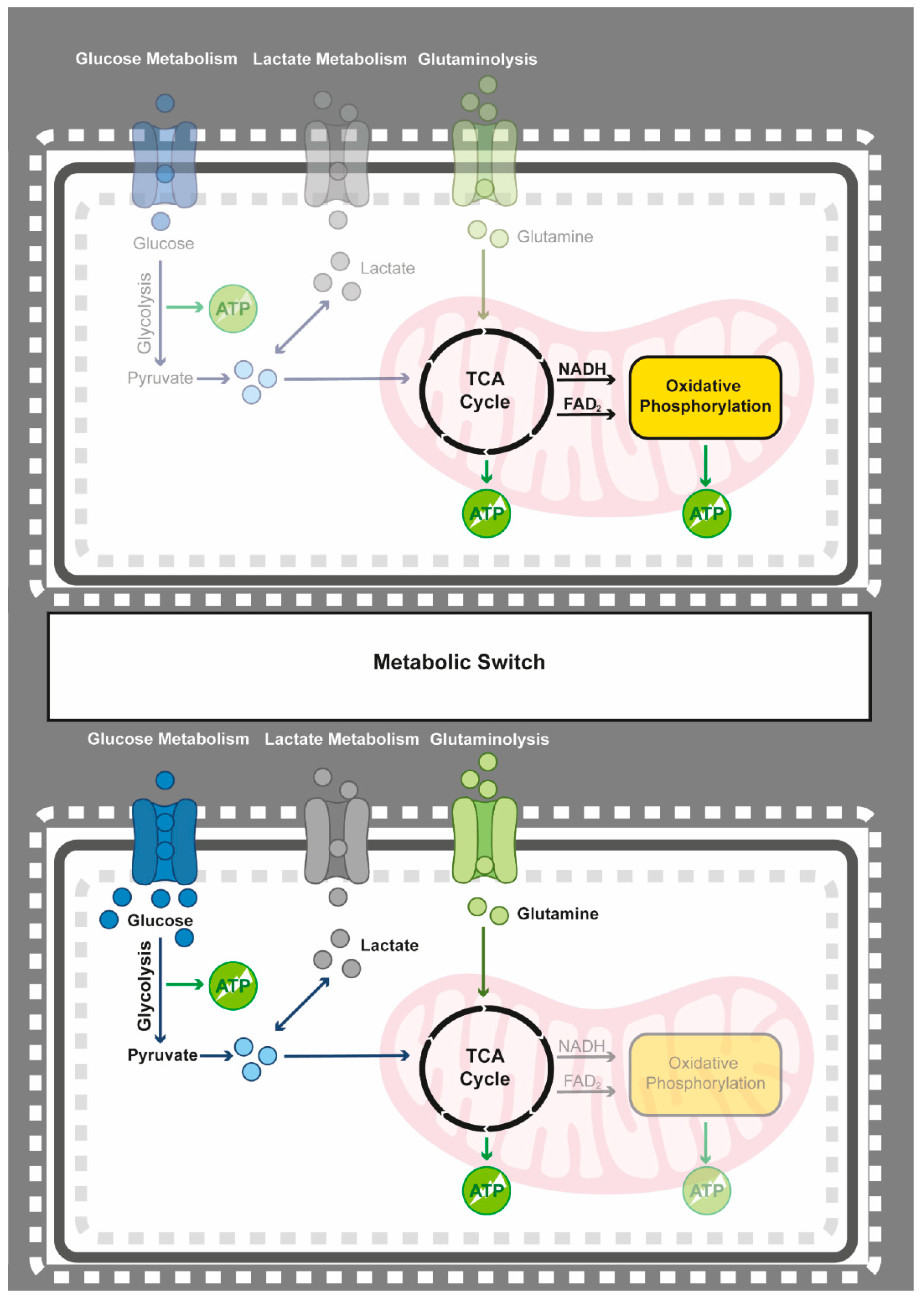
4.1. Epigenetics Teams up with the Metabolic Switch from Oxidative Phosphorylation to Glycolysis
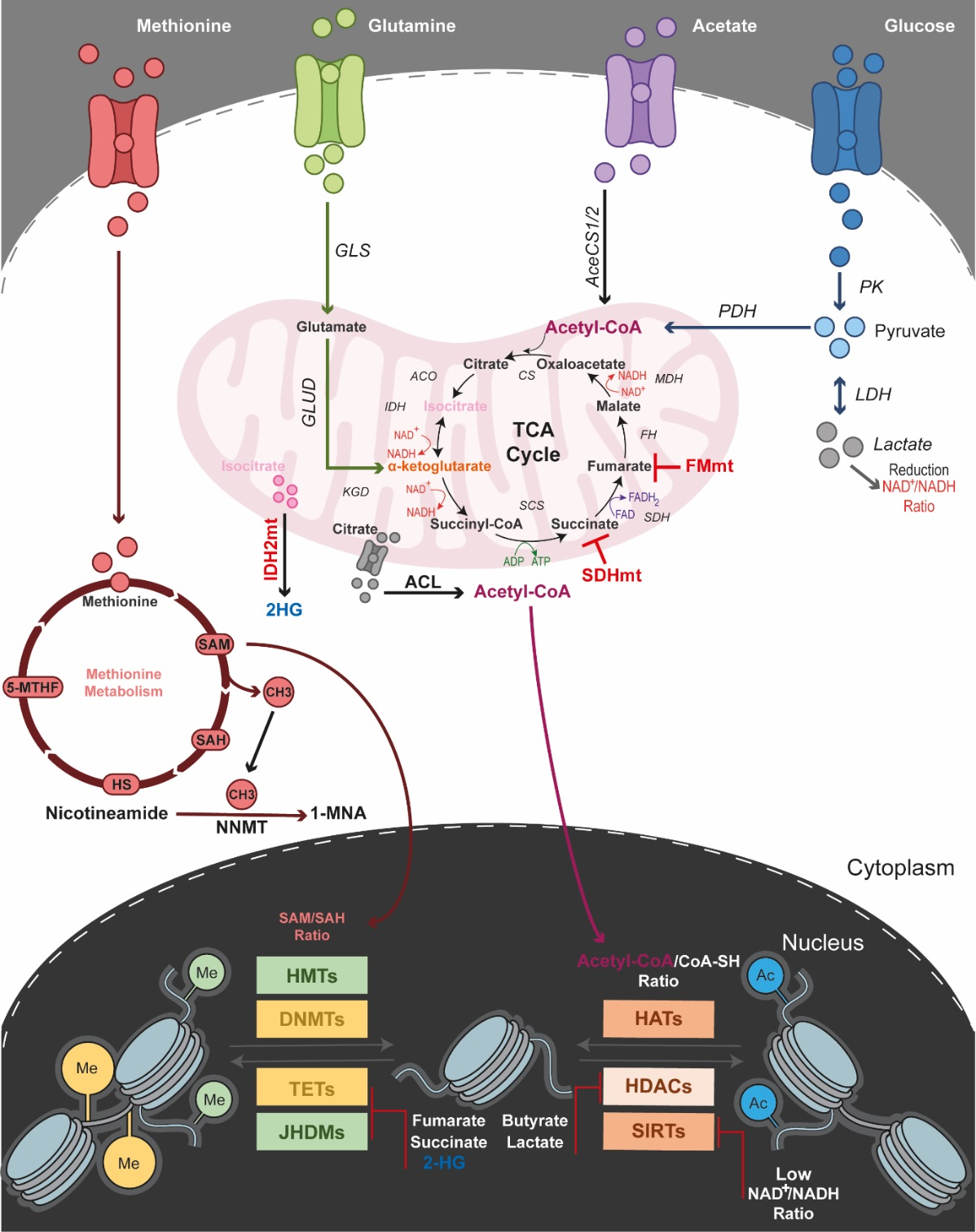
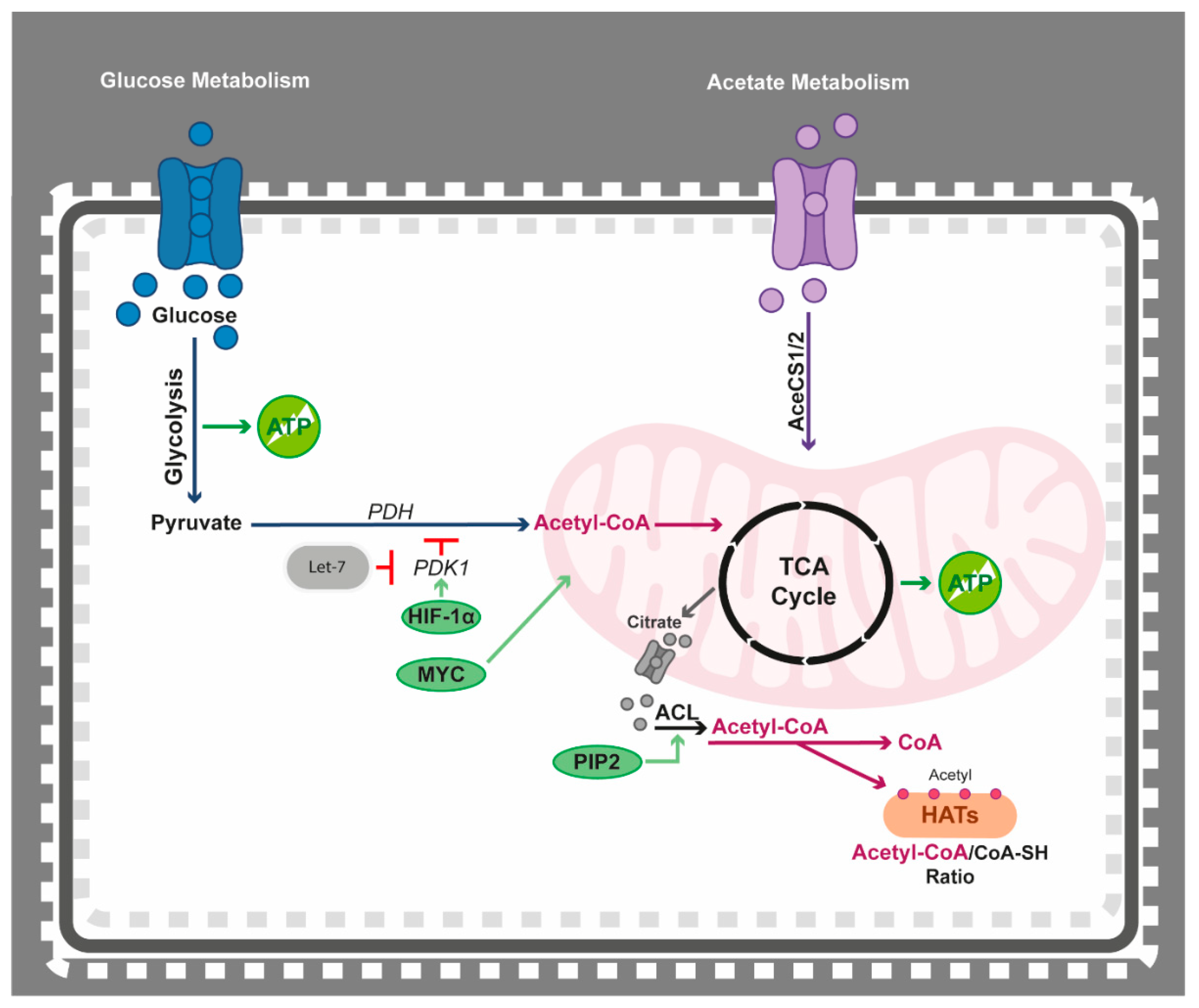
4.2. Epigenetics Teams up with Acetyl-CoA
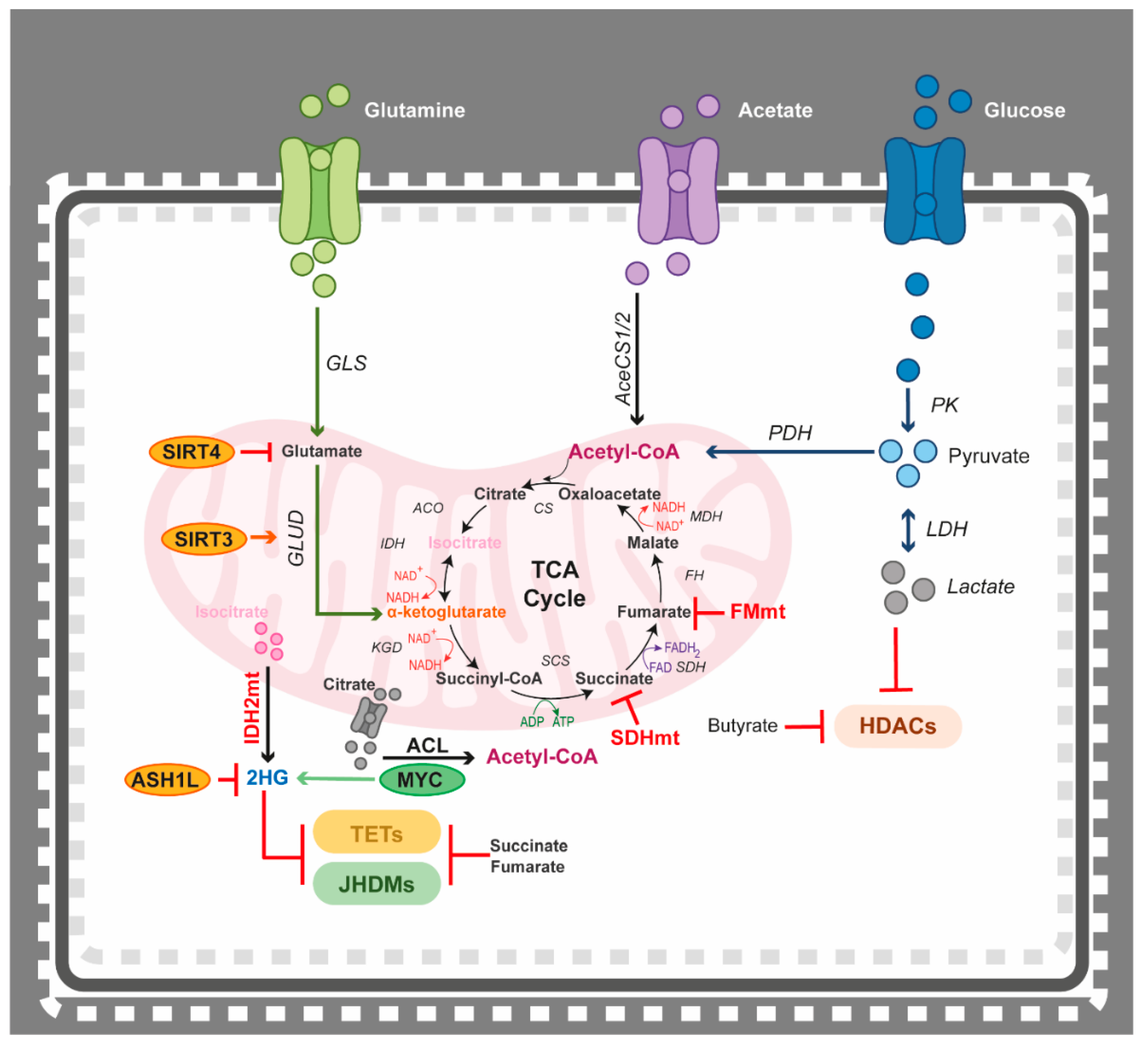
4.3. Epigenetics Teams up with the Tricarboxylic Acid (TCA) Cycle
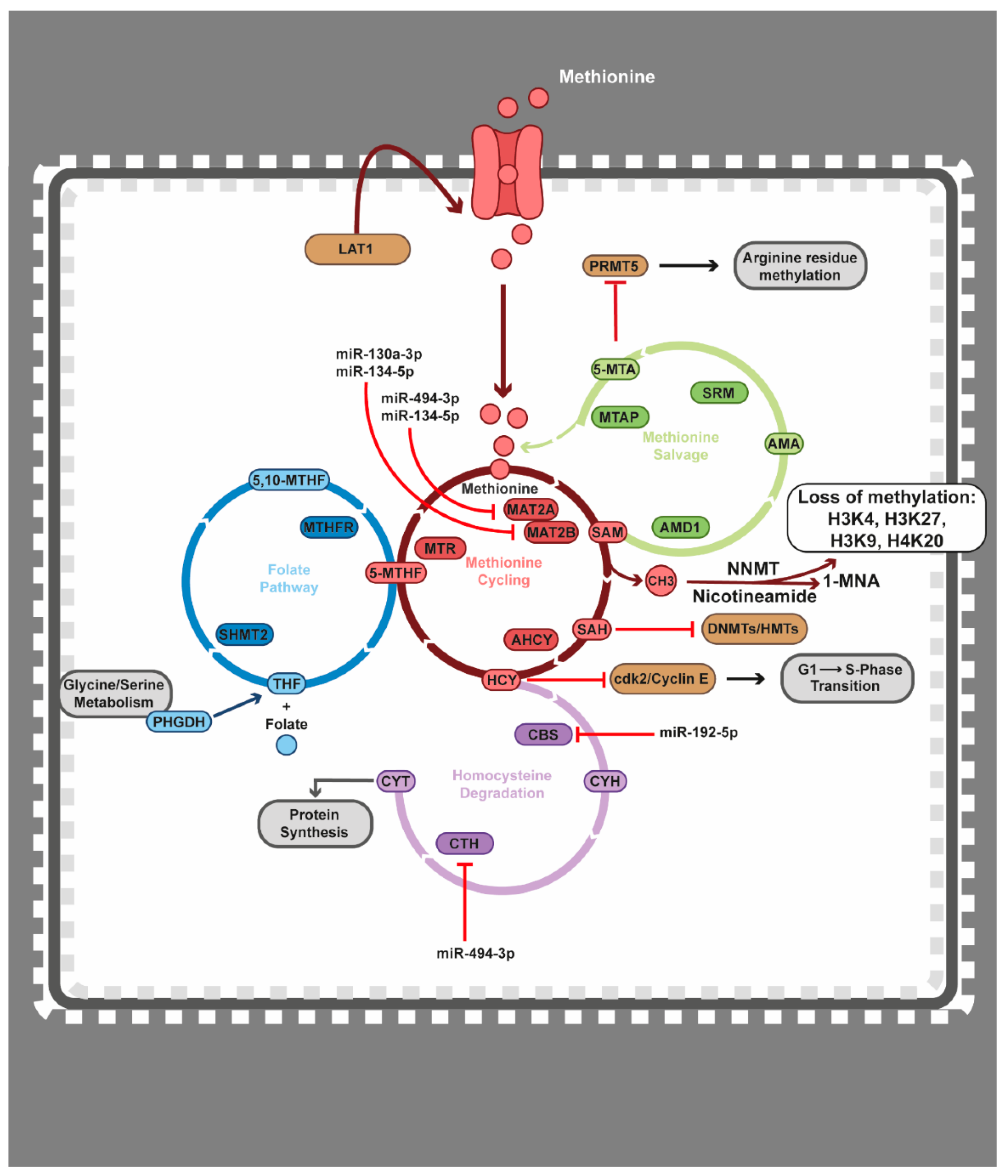
4.4. Epigenetics Teams up with the Methionine Cycle
5. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Robert, M.-F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2002, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Maiti, A.; Drohat, A.C. Thymine DNA Glycosylase Can Rapidly Excise 5-Formylcytosine and 5-Carboxylcytosine: POTENTIAL IMPLICATIONS FOR ACTIVE DEMETHYLATION OF CpG SITES. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ngo, T.T.M.; Yoo, J.; Dai, Q.; Zhang, Q.; He, C.; Aksimentiev, A.; Ha, T. Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat. Commun. 2016, 7, 10813. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Song, C.-X.; Du, T.; Jin, F.; Selvaraj, S.; Lee, A.Y.; Yen, C.-A.; Ye, Z.; Mao, S.-Q.; Wang, B.-A.; et al. 5mC Oxidation by Tet2 Modulates Enhancer Activity and Timing of Transcriptome Reprogramming during Differentiation. Mol. Cell 2014, 56, 286–297. [Google Scholar] [CrossRef]
- Duy, C.; Teater, M.; Garrett-Bakelman, F.E.; Lee, T.C.; Meydan, C.; Glass, J.L.; Li, M.; Hellmuth, J.C.; Mohammad, H.P.; Smitheman, K.N.; et al. Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML. Cancer Discov. 2019, 9, 872–889. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Milne, T.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. ALL-1 Is a Histone Methyltransferase that Assembles a Supercomplex of Proteins Involved in Transcriptional Regulation. Mol. Cell 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Wu, L.; Lee, S.Y.; Zhou, B.; Nguyen, U.T.; Muir, T.W.; Tan, S.; Dou, Y. ASH2L Regulates Ubiquitylation Signaling to MLL: Trans-Regulation of H3 K4 Methylation in Higher Eukaryotes. Mol. Cell 2013, 49, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mittal, A.; Reid, J.; Reich, S.; Gamblin, S.; Wilson, J.R. Evolving Catalytic Properties of the MLL Family SET Domain. Structure 2015, 23, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- De, S.; Shaknovich, R.; Riester, M.; Elemento, O.; Geng, H.; Kormaksson, M.; Jiang, Y.; Woolcock, B.; Johnson, N.; Polo, J.M.; et al. Aberration in DNA Methylation in B-Cell Lymphomas Has a Complex Origin and Increases with Disease Severity. PLoS Genet. 2013, 9, e1003137. [Google Scholar] [CrossRef]
- Chambwe, N.; Kormaksson, M.; Geng, H.; De, S.; Michor, F.; Johnson, N.A.; Morin, R.; Scott, D.W.; Godley, L.A.; Gascoyne, R.D.; et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 2014, 123, 1699–1708. [Google Scholar] [CrossRef]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef]
- Pan, H.; Renaud, L.; Chaligne, R.; Bloehdorn, J.; Tausch, E.; Mertens, D.; Fink, A.M.; Fischer, K.; Zhang, C.; Betel, D.; et al. Discovery of Candidate DNA Methylation Cancer Driver Genes. Cancer Discov. 2021, 11, 2266–2281. [Google Scholar] [CrossRef]
- Clozel, T.; Yang, S.; Elstrom, R.L.; Tam, W.; Martin, P.; Kormaksson, M.; Banerjee, S.; VasanthaKumar, A.; Culjkovic, B.; Scott, D.W.; et al. Mechanism-Based Epigenetic Chemosensitization Therapy of Diffuse Large B-Cell Lymphoma. Cancer Discov. 2013, 3, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Russler-Germain, D.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzén, S.; Hoglund, M.; Bullinger, L.; Döhner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated byDNMT3Amutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.L.; Hassane, D.; Wouters, B.J.; Kunimoto, H.; Avellino, R.; Garrett-Bakelman, F.E.; Guryanova, O.A.; Bowman, R.; Redlich, S.; Intlekofer, A.; et al. Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers. Cancer Discov. 2017, 7, 868–883. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.L.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.J.; et al. DNMT3AMutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Shen, Y.; Zhu, Y.-M.; Fan, X.; Shi, J.-Y.; Wang, Q.-R.; Yan, X.-J.; Gu, Z.-H.; Wang, Y.-Y.; Chen, B.; Jiang, C.-L.; et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood 2011, 118, 5593–5603. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: Mutations inRUNX1andDNMT3Aare associated with poor prognosis in T-ALL. Genes Chromosom. Cancer 2013, 52, 410–422. [Google Scholar] [CrossRef]
- Roller, A.; Grossmann, V.; Bacher, U.; Poetzinger, F.; Weissmann, S.; Nadarajah, N.; Boeck, L.; Kern, W.; Haferlach, T.; Schnittger, S.; et al. Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia 2013, 27, 1573–1578. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lodh, A.; Yevtodiyenko, A.; Liefwalker, D.; Li, H.; Felsher, D.W.; Van Riggelen, J. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt’s lymphoma. Oncotarget 2017, 8, 76898–76920. [Google Scholar] [CrossRef]
- Robaina, M.C.; Mazzoccoli, L.; Arruda, V.O.; Reis, F.R.D.S.; Apa, A.G.; de Rezende, L.M.M.; Klumb, C.E. Deregulation of DNMT1, DNMT3B and miR-29s in Burkitt lymphoma suggests novel contribution for disease pathogenesis. Exp. Mol. Pathol. 2015, 98, 200–207. [Google Scholar] [CrossRef]
- Amara, K.; Ziadi, S.; Hachana, M.; Soltani, N.; Korbi, S.; Trimeche, M. DNA methyltransferase DNMT3b protein overexpression as a prognostic factor in patients with diffuse large B-cell lymphomas. Cancer Sci. 2010, 101, 1722–1730. [Google Scholar] [CrossRef]
- Pawlyn, C.; Kaiser, M.F.; Heuck, C.; Melchor, L.; Wardell, C.; Murison, A.; Chavan, S.S.; Johnson, D.C.; Begum, D.B.; Dahir, N.M.; et al. The Spectrum and Clinical Impact of Epigenetic Modifier Mutations in Myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Moreau-Aubry, A.; Descamps, G.; Bougie, P.G.; Maïga, S.; Gaignard, A.; Chiron, D.; Ménoret, E.; le Gouill, S.; Moreau, P.; et al. Whole-exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J. Hematol. Oncol. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Fabris, S.; Pegoraro, V.; Ronchetti, D.; Mosca, L.; Deliliers, G.L.; Motta, V.; Bertazzi, P.A.; Baccarelli, A.; Neri, A. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009, 30, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Sive, J.I.; Feber, A.; Smith, D.; Quinn, J.; Beck, S.; Yong, K. Global hypomethylation in myeloma is associated with poor prognosis. Br. J. Haematol. 2015, 172, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Houde, C.; Li, Y.; Song, L.; Barton, K.; Zhang, Q.; Godwin, J.; Nand, S.; Toor, A.; Alkan, S.; Smadja, N.V.; et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 2004, 104, 3697–3704. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Atienza-Párraga, A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.; Brioli, A.; Mirabella, F.; Wardell, C.; Melchor, L.; Davies, F.; Morgan, G. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013, 122, 219–226. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.; Chiecchio, L.; Smith, E.M.; Boyd, K.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef]
- Heuck, C.J.; Mehta, J.; Bhagat, T.; Gundabolu, K.; Yu, Y.; Khan, S.; Chrysofakis, G.; Schinke, C.; Tariman, J.D.; Vickrey, E.; et al. Myeloma Is Characterized by Stage-Specific Alterations in DNA Methylation That Occur Early during Myelomagenesis. J. Immunol. 2013, 190, 2966–2975. [Google Scholar] [CrossRef]
- Agirre, X.; Castellano, G.; Pascual, M.; Heath, S.; Kulis, M.; Segura, V.; Bergmann, A.; Esteve-Codina, A.; Merkel, A.; Raineri, E.; et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015, 25, 478–487. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Ashby, C.; Tytarenko, R.; Bauer, M.; Wang, Y.; Deshpande, S.; Den, J.; Schinke, C.; Zangari, M.; Thanendrarajan, S.; et al. The functional epigenetic landscape of aberrant gene expression in molecular subgroups of newly diagnosed multiple myeloma. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Alberge, J.-B.; Magrangeas, F.; Wagner, M.; Denié, S.; Guérin-Charbonnel, C.; Campion, L.; Attal, M.; Avet-Loiseau, H.; Carell, T.; Moreau, P.; et al. DNA hydroxymethylation is associated with disease severity and persists at enhancers of oncogenic regions in multiple myeloma. Clin. Epigenetics 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Chatonnet, F.; Pignarre, A.; Sérandour, A.A.; Caron, G.; Avner, S.; Robert, N.; Kassambara, A.; Laurent, A.; Bizot, M.; Agirre, X.; et al. The hydroxymethylome of multiple myeloma identifies FAM72D as a 1q21 marker linked to proliferation. Haematologica 2019, 105, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Chou, S.-C.; Liu, C.-Y.; Chen, C.-Y.; Hou, H.-A.; Kuo, Y.-Y.; Lee, M.-C.; Ko, B.-S.; Tang, J.-L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Kern, W.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2011, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; Van Hoogen, P.; Van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.; Plo, I.; Wagner-Ballon, O.; Cruzeiro, M.D.; Delhommeau, F.; Arnulf, B.; Stern, M.-H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Béguelin, W.; Fontan, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation and promotes B-cell lymphomagenesis. Cancer Discov. 2018, 8, CD-18-0657-18. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 Mutations Are Frequent Genetic Alterations in Acute Myeloid Leukemia and Confer Adverse Prognosis in Cytogenetically Normal Acute Myeloid Leukemia With NPM1 Mutation Without FLT3 Internal Tandem Duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Abbas, S.; Lugthart, S.; Kavelaars, F.; Schelen, A.; Koenders, J.; Zeilemaker, A.; Van Putten, W.J.L.; Rijneveld, A.; Löwenberg, B.; Valk, P. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood 2010, 116, 2122–2126. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Ward, P.; Cross, J.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(−)-2-hydroxyglutarate production. Oncogene 2011, 31, 2491–2498. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.; Hillringhaus, L.; Bagg, E.; Rose, N.; Leung, I.; Li, X.S.; Woon, E.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Akalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA Hydroxymethylation Profiling Reveals that WT1 Mutations Result in Loss of TET2 Function in Acute Myeloid Leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Gupta, C.; Gabdoulline, R.; Borchert, N.M.; Goparaju, R.; Kaulfuss, S.; Görlich, K.; Schottmann, R.; Othman, B.; Welzenbach, J.; et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica 2020, 106, 565–573. [Google Scholar] [CrossRef]
- Dupéré-Richer, D.; Licht, J.D. Epigenetic regulatory mutations and epigenetic therapy for multiple myeloma. Curr. Opin. Hematol. 2017, 24, 336–344. [Google Scholar] [CrossRef]
- der Poel, S.Z.-V.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef]
- Gu, Y.; Nakamura, T.; Alder, H.; Prasad, R.; Canaani, O.; Cimino, G.; Croce, C. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 1992, 71, 701–708. [Google Scholar] [CrossRef]
- McCabe, N.R.; Burnett, R.C.; Gill, H.J.; Thirman, M.J.; Mbangkollo, D.; Kipiniak, M.; van Melle, E.; der Poel, S.Z.-V.; Rowley, J.D.; Diaz, M.O. Cloning of cDNAs of the MLL gene that detect DNA rearrangements and altered RNA transcripts in human leukemic cells with 11q23 translocations. Proc. Natl. Acad. Sci. USA 1992, 89, 11794–11798. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Gröger, D.; Park, T.S.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2017, 32, 273–284. [Google Scholar] [CrossRef]
- Erfurth, F.; Hemenway, C.S.; De Erkenez, A.C.; Domer, P.H. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia 2003, 18, 92–102. [Google Scholar] [CrossRef]
- Zeisig, D.T.; Bittner, C.B.; Zeisig, B.B.; García-Cuéllar, M.-P.; Hess, J.L.; Slany, R.K. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 2005, 24, 5525–5532. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.-P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Shilatifard, A.; Lane, W.S.; Jackson, K.W.; Conaway, R.C.; Conaway, J. An RNA Polymerase II Elongation Factor Encoded by the Human ELL Gene. Science 1996, 271, 1873–1876. [Google Scholar] [CrossRef]
- Hetzner, K.; Garcia-Cuellar, M.-P.; Büttner, C.; Slany, R.K. The interaction of ENL with PAF1 mitigates polycomb silencing and facilitates murine leukemogenesis. Blood 2018, 131, 662–673. [Google Scholar] [CrossRef] [PubMed]
- García-Cuéllar, M.P.; Zilles, O.; Schreiner, S.A.; Birke, M.; Winkler, T.H.; Slany, R.K. The ENL moiety of the childhood leukemia-associated MLL–ENL oncoprotein recruits human Polycomb 3. Oncogene 2001, 20, 411–419. [Google Scholar] [CrossRef]
- Lavau, C.; Du, C.; Thirman, M.; Zeleznik-Le, N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000, 19, 4655–4664. [Google Scholar] [CrossRef]
- Au, Y.Z.; Gu, M.; De Braekeleer, E.; Gozdecka, M.; Aspris, D.; Tarumoto, Y.; Cooper, J.; Yu, J.; Ong, S.H.; Chen, X.; et al. KAT7 is a genetic vulnerability of acute myeloid leukemias driven by MLL rearrangements. Leukemia 2020, 35, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.; Armstrong, S.A. Targeting Epigenetic Programs in MLL-Rearranged Leukemias. Hematology 2011, 2011, 354–360. [Google Scholar] [CrossRef]
- Xu, X.; Schneider, B. Therapeutic targeting potential of chromatin-associated proteins in MLL-rearranged acute leukemia. Cell. Oncol. 2018, 42, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Li, T.; Chen, Y.; Zhai, X. Nucleoside and Non-Nucleoside DOT1L Inhibitors: Dawn of MLLrearranged Leukemia. Mini-Rev. Med. Chem. 2021, 21, 1337–1350. [Google Scholar] [CrossRef]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K. Cytogenetic, Molecular Genetic, and Clinical Characteristics of Acute Myeloid Leukemia With a Complex Karyotype. Semin. Oncol. 2008, 35, 365–377. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Bullinger, L.; Basu, E.; Schlis, K.; Silverman, L.B.; Döhner, K.; Armstrong, S.A. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia 2012, 26, 1881–1883. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, C.M.; Nordlund, J.; Ekman, D.; Johansson, A.; Moghadam, B.T.; Raine, A.; Övernäs, E.; Dahlberg, J.; Wahlberg, P.; Henriksson, N.; et al. The Mutational Landscape in Pediatric Acute Lymphoblastic Leukemia Deciphered by Whole Genome Sequencing. Hum. Mutat. 2014, 36, 118–128. [Google Scholar] [CrossRef]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2014, 6, 2754–2766. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nat. Cell Biol. 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Herz, H.-M.; Hu, D.; Shilatifard, A. Enhancer Malfunction in Cancer. Mol. Cell 2014, 53, 859–866. [Google Scholar] [CrossRef]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef]
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.-E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 2015, 21, 1190–1198. [Google Scholar] [CrossRef]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Kroeze, L.; Nikoloski, G.; Da Silva-Coelho, P.; Van Hoogen, P.; Stevens-Linders, E.; Kuiper, R.P.; Schnittger, S.; Haferlach, T.; Pahl, H.L.; Van Der Reijden, B.A.; et al. Genetic defects in PRC2 components other than EZH2 are not common in myeloid malignancies. Blood 2012, 119, 1318–1319. [Google Scholar] [CrossRef]
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2014, 263, 22–35. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Ernst, T.; Pflug, A.; Rinke, J.; Ernst, J.; Bierbach, U.; Beck, J.F.; Hochhaus, A.; Gruhn, B. A somatic EZH2 mutation in childhood acute myeloid leukemia. Leukemia 2012, 26, 1701–1703. [Google Scholar] [CrossRef][Green Version]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; Van Der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; De Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; Da Ros, V.G.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–302. [Google Scholar] [CrossRef]
- Score, J.; Hidalgo-Curtis, C.; Jones, A.V.; Winkelmann, N.; Skinner, A.; Ward, D.; Zoi, K.; Ernst, T.; Stegelmann, F.; Döhner, K.; et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012, 119, 1208–1213. [Google Scholar] [CrossRef]
- Simon, C.; Chagraoui, J.; Krosl, J.; Gendron, P.; Wilhelm, B.; Lemieux, S.; Boucher, G.; Chagnon, P.; Drouin, S.; Lambert, R.; et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012, 26, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kohlmann, A.; Zenger, M.; Schindela, S.; Eder, C.; Weissmann, S.; Schnittger, S.; Kern, W.; Muller, M.C.; Hochhaus, A.; et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 2011, 25, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Bödör, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.-Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2013, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.; Love, C.L.; Waldrop, A.; Leppä, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef]
- Yap, D.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.-W.G.; Moradian, A.; Morin, R.D.; Mungall, A.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef]
- McCabe, M.T.; Graves, A.P.; Ganji, G.; Diaz, E.; Halsey, W.S.; Jiang, Y.; Smitheman, K.N.; Ott, H.M.; Pappalardi, M.B.; Allen, K.E.; et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Béguelin, W.; Teater, M.; Gearhart, M.; Fernández, M.T.C.; Goldstein, R.L.; Cárdenas, M.G.; Hatzi, K.; Rosen, M.; Shen, H.; Corcoran, C.M.; et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016, 30, 197–213. [Google Scholar] [CrossRef]
- Herviou, L.; Jourdan, M.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. EZH2 is overexpressed in transitional preplasmablasts and is involved in human plasma cell differentiation. Leukemia 2019, 33, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Croonquist, P.A.; Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Alzrigat, M.; Atienza-Párraga, A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Österborg, A.; Brown, P.; Ma, A.; Jin, J.; et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Kalushkova, A.; Fryknäs, M.; Lemaire, M.; Fristedt, C.; Agarwal, P.; Eriksson, M.; Deleu, S.; Atadja, P.; Österborg, A.; Nilsson, K.; et al. Polycomb Target Genes Are Silenced in Multiple Myeloma. PLoS ONE 2010, 5, e11483. [Google Scholar] [CrossRef] [PubMed]
- Herviou, L.; Kassambara, A.; Boireau, S.; Robert, N.; Requirand, G.; Müller-Tidow, C.; Vincent, L.; Seckinger, A.; Goldschmidt, H.; Cartron, G.; et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin. Epigenetics 2018, 10, 121. [Google Scholar] [CrossRef]
- Harding, T.; Swanson, J.; Van Ness, B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9, 21930–21942. [Google Scholar] [CrossRef]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol. Cancer Ther. 2015, 15, 287–298. [Google Scholar] [CrossRef]
- Pawlyn, C.; Bright, M.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef]
- Zeng, D.; Liu, M.; Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 2016, 8, 3396–3411. [Google Scholar] [CrossRef]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.-H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef]
- Rizk, M.; Rizq, O.; Oshima, M.; Nakajima-Takagi, Y.; Koide, S.; Saraya, A.; Isshiki, Y.; Chiba, T.; Yamazaki, S.; Ma, A.; et al. Akt inhibition synergizes with polycomb repressive complex 2 inhibition in the treatment of multiple myeloma. Cancer Sci. 2019, 110, 3695–3707. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tang, H.; Wang, K.; Zheng, Y.; Feng, J.; Dong, H.; Jin, Y.; Cao, C.; Chen, X.; Gao, G. Pharmacological inhibition of EZH2 combined with DNA-damaging agents interferes with the DNA damage response in MM cells. Mol. Med. Rep. 2019, 19, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Du, C.; Ma, X.; Sui, W.; Yu, Z.; Liu, L.; Zhao, L.; Li, Z.; Xu, J.; Wei, X.; et al. Polycomb-like Protein 3 Induces Proliferation and Drug Resistance in Multiple Myeloma and Is Regulated by miRNA-15a. Mol. Cancer Res. 2020, 18, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Côté, S.; Momparler, L.F.; Idaghdour, Y. Inhibition of DNA and Histone Methylation by 5-Aza-2′-Deoxycytidine (Decitabine) and 3-Deazaneplanocin-A on Antineoplastic Action and Gene Expression in Myeloid Leukemic Cells. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Cerveira, N.; Correia, C.; Dória, S.; Bizarro, S.D.C.; Rocha, P.; Gomes, P.; Torres, L.; Norton, L.; Borges, B.S.; Castedo, S.; et al. Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 2003, 17, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nature 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Oyer, J.A.; Huang, X.; Zheng, Y.; Shim, J.; Ezponda, T.; Carpenter, Z.; Allegretta, M.; Okot-Kotber, C.I.; Patel, J.P.; Melnick, A.; et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2013, 28, 198–201. [Google Scholar] [CrossRef]
- Keats, J.; Reiman, T.; Maxwell, C.; Taylor, B.J.; Larratt, L.M.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [Google Scholar] [CrossRef]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, E.; Popovic, R.; Min, D.-J.; Sweet, S.; Thomas, P.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef]
- An The Cancer Genome Atlas Research Network Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [PubMed]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [PubMed]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Popovic, R.; Zheng, Y.; Nabet, B.; Will, M.C.; Small, E.C.; Occhionorelli, M.; Tonon, M.G.; Keats, J.J.; Kelleher, N.L.; et al. Loss of the Histone Demethylase UTX Contributes to Multiple Myeloma and Sensitizes Cells to EZH2 Inhibitors. Blood 2014, 124, 611. [Google Scholar] [CrossRef]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.-P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef]
- Sze, C.C.; Shilatifard, A. MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026427. [Google Scholar] [CrossRef]
- Wei, X.; Calvo-Vidal, M.N.; Chen, S.; Wu, G.; Revuelta, M.V.; Sun, J.; Zhang, J.; Walsh, M.F.; Nichols, K.E.; Joseph, V.; et al. Germline Lysine-Specific Demethylase 1 (LSD1/KDM1A) Mutations Confer Susceptibility to Multiple Myeloma. Cancer Res. 2018, 78, 2747–2759. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.-T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Soncini, D.; Orecchioni, S.; Talarico, G.; Minetto, P.; Guolo, F.; Retali, V.; Colombo, N.; Carminati, E.; Clavio, M.; et al. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica 2017, 103, 80–90. [Google Scholar] [CrossRef]
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227. [Google Scholar] [CrossRef]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef]
- Hansen, V.L.; Coleman, M.; Elkins, S.; Letzer, J.P.; Levy, M.Y.; Seneviratne, L.; Rine, J.; White, M.; Kuriakose, E.T. An Expanded Treatment Protocol of Panobinostat Plus Bortezomib and Dexamethasone in Patients With Previously Treated Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 400–407.e1. [Google Scholar] [CrossRef]
- Han, Z.; Rosen, S.T.; Querfeld, C. Targeting microRNA in hematologic malignancies. Curr. Opin. Oncol. 2020, 32, 535–544. [Google Scholar] [CrossRef]
- Katsaraki, K.; Karousi, P.; Artemaki, P.; Scorilas, A.; Pappa, V.; Kontos, C.; Papageorgiou, S. MicroRNAs: Tiny Regulators of Gene Expression with Pivotal Roles in Normal B-Cell Development and B-Cell Chronic Lymphocytic Leukemia. Cancers 2021, 13, 593. [Google Scholar] [CrossRef]
- Marton, S.; Garcia, M.R.; Robello, C.; Persson, H.; Trajtenberg, F.; Pritsch, O.; Rovira, C.; Naya, H.; Dighiero, G.; Cayota, A. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 2007, 22, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zanette, D.; Rivadavia, F.; Molfetta, G.; Barbuzano, F.; Proto-Siqueira, R.; Falcão, R.; Zago, M.; Silva, W., Jr. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz. J. Med. Biol. Res. 2007, 40, 1435–1440. [Google Scholar] [CrossRef]
- Alzrigat, M.; Atienza-Párraga, A.; Agarwal, P.; Zureigat, H.; Österborg, A.; Nahi, H.; Ma, A.; Jin, J.; Nilsson, K.; Öberg, F.; et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 2016, 8, 10213–10224. [Google Scholar] [CrossRef]
- Fulci, V.; Chiaretti, S.; Goldoni, M.; Azzalin, G.; Carucci, N.; Tavolaro, S.; Castellano, L.; Magrelli, A.; Citarella, F.; Messina, M.; et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007, 109, 4944–4951. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, B.; Meuleman, N.; Haibe-Kains, B.; Saussoy, P.; Neste, E.V.D.; Michaux, L.; Heimann, P.; Martiat, P.; Bron, D.; Lagneaux, L. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 2009, 113, 5237–5245. [Google Scholar] [CrossRef]
- Rossi, S.; Shimizu, M.; Barbarotto, E.; Nicoloso, M.S.; Dimitri, F.; Sampath, D.; Fabbri, M.; Lerner, S.; Barron, L.L.; Rassenti, L.Z.; et al. microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood 2010, 116, 945–952. [Google Scholar] [CrossRef]
- Gutierrez, N.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; Rivas, J.D.L.; Ticona, F.V.; Fermiñán, E.; Martín-Jiménez, P.; Chillon, M.C.; Risueño, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef]
- Chi, J.; Ballabio, E.; Chen, X.-H.; Kušec, R.; Taylor, S.; Hay, D.; Tramonti, D.; Saunders, N.J.; Littlewood, T.; Pezzella, F.; et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct 2011, 6, 23. [Google Scholar] [CrossRef]
- Kassambara, A.; Jourdan, M.; Bruyer, A.; Robert, N.; Pantesco, V.; Elemento, O.; Klein, B.; Moreaux, J. Global miRNA expression analysis identifies novel key regulators of plasma cell differentiation and malignant plasma cell. Nucleic Acids Res. 2017, 45, 5639–5652. [Google Scholar] [CrossRef]
- Eis, P.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Fava, P.; Bergallo, M.; Astrua, C.; Brizio, M.; Galliano, I.; Montanari, P.; Daprà, V.; Novelli, M.; Savoia, P.; Quaglino, P.; et al. miR-155 expression in Primary Cutaneous T-Cell Lymphomas (CTCL). J. Eur. Acad. Dermatol. Venereol. 2016, 31, e27–e29. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Barzilai, A.; Gorovitz, B.; Hirshberg, A.; Amariglio, N.; Jacob-Hirsch, J.; Maron, L.; Feinmesser, M.; Hodak, E. miR-155 is involved in tumor progression of mycosis fungoides. Exp. Dermatol. 2013, 22, 431–433. [Google Scholar] [CrossRef]
- Gerloff, D.; Grundler, R.; Wurm, A.A.; Bräuer-Hartmann, D.; Katzerke, C.; Hartmann, J.-U.; Madan, V.; Müller-Tidow, C.; Duyster, J.; Tenen, D.G.; et al. NF-κB/STAT5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia 2014, 29, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Palma, C.A.; Al Sheikha, D.; Lim, T.K.; Bryant, A.; Vu, T.T.; Jayaswal, V.; Ma, D.D.F. MicroRNA-155 as an inducer of apoptosis and cell differentiation in Acute Myeloid Leukaemia. Mol. Cancer 2014, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Ferrajoli, A.; Shanafelt, T.D.; Ivan, C.; Shimizu, M.; Rabe, K.G.; Nouraee, N.; Ikuo, M.; Ghosh, A.K.; Lerner, S.; Rassenti, L.Z.; et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood 2013, 122, 1891–1899. [Google Scholar] [CrossRef]
- Lawrie, C.H.; Soneji, S.; Marafioti, T.; Cooper, C.D.; Palazzo, S.; Paterson, J.C.; Cattan, H.; Enver, T.; Mager, R.; Boultwood, J.; et al. Microrna expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int. J. Cancer 2007, 121, 1156–1161. [Google Scholar] [CrossRef]
- Mi, S.; Li, Z.; Chen, P.; He, C.; Cao, D.; Elkahloun, A.; Lu, J.; Pelloso, L.A.; Wunderlich, M.; Huang, H.; et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3710–3715. [Google Scholar] [CrossRef]
- Poláková, K.M.; Lopotová, T.; Klamová, H.; Burda, P.; Trněný, M.; Stopka, T.; Moravcová, J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol. Cancer 2011, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Pichiorri, F.; Palumbo, T.; Visentini, M.; Aqeilan, R.; Cimmino, A.; Wang, H.; Sun, H.; Volinia, S.; Alder, H.; et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 2007, 26, 4148–4157. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Kitade, Y.; Kinoshita, T.; Naoe, T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007, 98, 1914–1920. [Google Scholar] [CrossRef]
- Chen, C.-Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, S.; Skoulakis, S.; Molloy, G.; Chaplin, T.; Dixon-McIver, A.; Young, B.D. MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 2007, 21, 912–916. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, H.; Fang, Z.; Fan, Y.; Liu, X.; Zhang, Y.; Rui, S.; Chen, Y.; Hong, L.; Zhang, J.J.G.M.; et al. MiR-204 acts as a potential therapeutic target in acute myeloid leukemia by increasing BIRC6-mediated apoptosis. BMB Rep. 2018, 51, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, Z.; Lu, R.; Zhao, H.; Gong, T.; Liu, D.; Hong, L.; Ma, J.; Zhang, M. MicroRNA-204 Potentiates the Sensitivity of Acute Myeloid Leukemia Cells to Arsenic Trioxide. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2019, 27, 1035–1042. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, P.; Chen, Y.; Chen, J.; Ma, R.; Sun, L. Overexpression of microRNA-125b inhibits human acute myeloid leukemia cells invasion, proliferation and promotes cells apoptosis by targeting NF-κB signaling pathway. Biochem. Biophys. Res. Commun. 2017, 488, 60–66. [Google Scholar] [CrossRef]
- Shen, M.Y.; Wang, Y.; Cui, S.Y.; Wu, X.L.; Guo, Y.; Xu, R.R. MicroRNA-125a Regulates Proliferation and Apoptosis of Acute Myeloid Leukemia through Targeting NF-ΚB Pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3594–3601. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Pierce, S.; Bueso-Ramos, C.; Calin, G.; Boyano-Adánez, M.D.C.; García-Manero, G. Overexpression of miR-125a in Myelodysplastic Syndrome CD34+ Cells Modulates NF-κB Activation and Enhances Erythroid Differentiation Arrest. PLoS ONE 2014, 9, e93404. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Gerace, D.; Bianco, O.; Musolino, C. The metabolomic signature of hematologic malignancies. Leuk. Res. 2016, 49, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Pavlova, N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Xu, X. Knockdown of LncRNA-UCA1 suppresses chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA-125a/hexokinase 2 pathway. J. Cell. Biochem. 2018, 119, 6296–6308. [Google Scholar] [CrossRef] [PubMed]
- English, S.G.; Hadj-Moussa, H.; Storey, K.B. MicroRNAs regulate survival in oxygen-deprived environments. J. Exp. Biol. 2018, 221, jeb.190579. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kitadate, A.; Abe, F.; Takahashi, N.; Tagawa, H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2018, 2, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, P.; Vandsemb, E.N.; Elsaadi, S.; Røst, L.M.; Yang, R.; Hjort, M.A.; Andreassen, T.; Misund, K.; Slørdahl, T.S.; Rø, T.B.; et al. Phosphatase of regenerating liver-3 regulates cancer cell metabolism in multiple myeloma. FASEB J. 2021, 35, e21344. [Google Scholar] [CrossRef]
- Vandsemb, E.N.; Rye, M.B.; Steiro, I.J.; Elsaadi, S.; Rø, T.B.; Slørdahl, T.S.; Sponaas, A.; Børset, M.; Abdollahi, P. PRL-3 induces a positive signaling circuit between glycolysis and activation of STAT1/2. FEBS J. 2021. [Google Scholar] [CrossRef]
- Caillot, M.; Bourgeais, J.; Dakik, H.; Costé, E.; Mazure, N.M.; Lelievre, E.; Coqueret, O.; Hérault, O.; Mazurier, F.; Sola, B. Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Zhan, G.; Huang, A.; Sun, Y.; Wen, S.; Yang, J.; Lu, W.-H.; Xu, R.-H.; Li, J.; Li, Y.; et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 2017, 31, 2143–2150. [Google Scholar] [CrossRef]
- Ryu, M.J.; Han, J.; Kim, S.J.; Lee, M.J.; Ju, X.; Lee, Y.L.; Son, J.H.; Cui, J.; Jang, Y.; Chung, W.; et al. PTEN/AKT signaling mediates chemoresistance in refractory acute myeloid leukemia through enhanced glycolysis. Oncol. Rep. 2019, 42, 2149–2158. [Google Scholar] [CrossRef]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.; Siska, P.J.; Macintyre, A.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef]
- Karagianni, P.; Giannouli, S.; Voulgarelis, M. From the (Epi)Genome to Metabolism and Vice Versa; Examples from Hematologic Malignancy. Int. J. Mol. Sci. 2021, 22, 6321. [Google Scholar] [CrossRef]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef]
- Cui, L.; Cheng, Z.; Liu, Y.; Dai, Y.; Pang, Y.; Jiao, Y.; Ke, X.; Cui, W.; Zhang, Q.; Shi, J.; et al. Overexpression of PDK2 and PDK3 reflects poor prognosis in acute myeloid leukemia. Cancer Gene Ther. 2018, 27, 15–21. [Google Scholar] [CrossRef]
- Weisberg, E.; Banerji, L.; Wright, R.D.; Barrett, R.; Ray, A.; Moreno, D.; Catley, L.; Jiang, J.; Hall-Meyers, E.; Sauveur-Michel, M.; et al. Potentiation of antileukemic therapies by the dual PI3K/PDK-1 inhibitor, BAG956: Effects on BCR-ABL– and mutant FLT3-expressing cells. Blood 2008, 111, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Somers, K.; Wen, V.W.; Middlemiss, S.M.C.; Osborne, B.; Forgham, H.; Jung, M.; Karsa, M.; Clifton, M.; Bongers, A.; Gao, J.; et al. A novel small molecule that kills a subset of MLL-rearranged leukemia cells by inducing mitochondrial dysfunction. Oncogene 2019, 38, 3824–3842. [Google Scholar] [CrossRef] [PubMed]
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br. J. Cancer 2016, 114, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-L.; Abudureheman, T.; Xia, J.; Chu, L.; Zhou, H.; Zheng, W.-W.; Zhou, N.; Shi, R.-Y.; Li, M.-H.; Zhu, J.-M.; et al. CDK9 Inhibitor Induces the Apoptosis of B-Cell Acute Lymphocytic Leukemia by Inhibiting c-Myc-Mediated Glycolytic Metabolism. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- De Rosa, V.; Monti, M.; Terlizzi, C.; Fonti, R.; Del Vecchio, S.; Iommelli, F. Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells. Int. J. Mol. Sci. 2019, 20, 3134. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Jimi, S.; Migita, K.; Takamatsu, Y.; Hara, S. Inhibition of glucose transporter 1 induces apoptosis and sensitizes multiple myeloma cells to conventional chemotherapeutic agents. Leuk. Res. 2015, 41, 103–110. [Google Scholar] [CrossRef]
- Åbacka, H.; Hansen, J.S.; Huang, P.; Venskutonytė, R.; Hyrenius-Wittsten, A.; Poli, G.; Tuccinardi, T.; Granchi, C.; Minutolo, F.; Hagström-Andersson, A.K.; et al. Targeting GLUT1 in acute myeloid leukemia to overcome cytarabine resistance. Haematologica 2020, 106, 1163–1166. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Wang, Y.-Y.; Zhao, A.; Xia, L.; Xie, G.; Su, M.; Zhao, L.; Liu, J.; Qu, C.; Wei, R.; et al. Enhanced Fructose Utilization Mediated by SLC2A5 Is a Unique Metabolic Feature of Acute Myeloid Leukemia with Therapeutic Potential. Cancer Cell 2016, 30, 779–791. [Google Scholar] [CrossRef]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Drive Proliferation in Acute Myeloid Leukemia via the Glycolytic Regulator PFKFB3. Cancer Res. 2019, 80, 937–949. [Google Scholar] [CrossRef]
- Hulleman, E.; Kazemier, K.M.; Holleman, A.; VanderWeele, D.; Rudin, C.; Broekhuis, M.J.C.; Evans, W.E.; Pieters, R.; Boer, M.D. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 2009, 113, 2014–2021. [Google Scholar] [CrossRef]
- Hanson, D.J.; Nakamura, S.; Amachi, R.; Hiasa, M.; Oda, A.; Tsuji, D.; Itoh, K.; Harada, T.; Horikawa, K.; Teramachi, J.; et al. Effective impairment of myeloma cells and their progenitors by blockade of monocarboxylate transportation. Oncotarget 2015, 6, 33568–33586. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hao, X.; Lai, X.; Liu, L.; Zhu, J.; Shao, H.; Huang, D.; Gu, H.; Zhang, T.; Yu, Z.; et al. Oxidative phosphorylation enhances the leukemogenic capacity and resistance to chemotherapy of B cell acute lymphoblastic leukemia. Sci. Adv. 2021, 7, eabd6280. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Diz, V.; Cao, B.; Lancho, O.; Chiles, E.; Alasadi, A.; Aleksandrova, M.; Luo, S.; Singh, A.; Tao, H.; Augeri, D.J.; et al. A novel and highly effective mitochondrial uncoupling drug in T-cell leukemia. Blood 2021. [Google Scholar] [CrossRef]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Gießl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Wang, J.; Ye, W.; Yan, X.; Guo, Q.; Ma, Q.; Lin, F.; Huang, J.; Jin, J. Low expression of ACLY associates with favorable prognosis in acute myeloid leukemia. J. Transl. Med. 2019, 17, 149. [Google Scholar] [CrossRef]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef]
- Kim, J.-A.; Yeom, Y.I. Metabolic Signaling to Epigenetic Alterations in Cancer. Biomol. Ther. 2018, 26, 69–80. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; He, J.; Wang, Z.; Yin, Z.; You, G.; Wang, Z.; Davis, R.E.; Lin, P.; Bergsagel, P.L.; et al. Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021, 33, 78–93.e7. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Miwa, H.; Shikami, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Tsunekawa-Imai, N.; Ishikawa, T.; Mizutani, M.; Horio, T.; Gotou, M.; et al. Leukemia cells demonstrate a different metabolic perturbation provoked by 2-deoxyglucose. Oncol. Rep. 2013, 29, 2053–2057. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travins, J.; Wang, F.; David, M.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DelaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D.; Paul, B.; Kang, Y. Metabolic alterations and the potential for targeting metabolic pathways in the treatment of multiple myeloma. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Dyczynski, M.; Vesterlund, M.; Björklund, A.-C.; Zachariadis, V.; Janssen, J.; Gallart-Ayala, H.; Daskalaki, E.; Wheelock, C.E.; Lehtiö, J.; Grandér, D.; et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R.; Matulis, S.M.; Wei, C.; Nooka, A.K.; Von Hollen, H.E.; Lonial, S.; Boise, L.; Shanmugam, M. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene 2015, 35, 3955–3964. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell 2017, 172, 90–105.e23. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Elo, L.L.; Karjalainen, R.; Öhman, T.; Hintsanen, P.; Nyman, T.A.; Heckman, C.A.; Aittokallio, T. Statistical detection of quantitative protein biomarkers provides insights into signaling networks deregulated in acute myeloid leukemia. Proteomics 2014, 14, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Nickerson, M.L.; Wei, M.-H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; et al. Mutations in the Fumarate Hydratase Gene Cause Hereditary Leiomyomatosis and Renal Cell Cancer in Families in North America. Am. J. Hum. Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef]
- Baysal, B.E. A Recurrent Stop-Codon Mutation in Succinate Dehydrogenase Subunit B Gene in Normal Peripheral Blood and Childhood T-Cell Acute Leukemia. PLoS ONE 2007, 2, e436. [Google Scholar] [CrossRef] [PubMed]
- Renella, R.; Carnevale, J.; Schneider, K.A.; Hornick, J.L.; Rana, H.Q.; Janeway, K.A. Exploring the association of succinate dehydrogenase complex mutations with lymphoid malignancies. Fam. Cancer 2014, 13, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The Effect of Replacement of Methionine by Homocystine on Survival of Malignant and Normal Adult Mammalian Cells in Culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.J.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2014, 29, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Maddocks, O.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Elsaadi, S.; Steiro, I.; Abdollahi, P.; Vandsemb, E.N.; Yang, R.; Slørdahl, T.S.; Rø, T.B.; Menu, E.; Sponaas, A.-M.; Børset, M. Targeting phosphoglycerate dehydrogenase in multiple myeloma. Exp. Hematol. Oncol. 2021, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Corbet, C.; Pinto, A.; Rubio, L.I.; Martherus, R.; Bol, V.; Drozak, X.; Grégoire, V.; Riant, O.; Feron, O. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 2015, 7, 1765–1776. [Google Scholar] [CrossRef]
- Białopiotrowicz, E.; Noyszewska-Kania, M.; Kachamakova-Trojanowska, N.; Łoboda, A.; Cybulska, M.; Grochowska, A.; Kopczyński, M.; Mikula, M.; Prochorec-Sobieszek, M.; Firczuk, M.; et al. Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers 2020, 12, 580. [Google Scholar] [CrossRef]
- Zarou, M.M.; Vazquez, A.; Helgason, G.V. Folate metabolism: A re-emerging therapeutic target in haematological cancers. Leukemia 2021, 35, 1539–1551. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Lu, C.; Thompson, C.B. Metabolic Regulation of Epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Järviaho, T.; Hurme-Niiranen, A.; Soini, H.K.; Niinimäki, R.; Möttönen, M.; Savolainen, E.; Hinttala, R.; Harila-Saari, A.; Uusimaa, J. Novel non-neutral mitochondrial DNA mutations found in childhood acute lymphoblastic leukemia. Clin. Genet. 2017, 93, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Xu, N.; Zhou, X.; Huang, J.; Wan-Er, W.; Chen, C.; Liang, L.; Liu, Q.; Xiaoli, L. PKM2 Mediates Chronic Myeloid Leukemia Imatinib Resistance By Regulating Glycolysis Energy Metabolism. Blood 2018, 132, 1724. [Google Scholar] [CrossRef]
- Evens, A.M.; Schumacker, P.T.; Helenowski, I.B.; Singh, A.T.K.; Dokic, D.; Keswani, A.; Kordeluk, E.; Raji, A.; Winter, J.N.; Jovanovic, B.D.; et al. Hypoxia inducible factor-alpha activation in lymphoma and relationship to the thioredoxin family. Br. J. Haematol. 2008, 141, 676–680. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.I.; Pezzella, F.; Sivridis, E.; Turley, H.; Harris, A.L.; Gatter, K.C. Phosphorylated VEGFR2/KDR receptors are widely expressed in B-cell non-Hodgkin’s lymphomas and correlate with hypoxia inducible factor activation. Hematol. Oncol. 2008, 26, 219–224. [Google Scholar] [CrossRef]
- Wellmann, S.; Guschmann, M.; Griethe, W.; Eckert, C.; Stackelberg, A.; Lottaz, C.; Moderegger, E.; Einsiedel, H.G.; Eckardt, K.-U.; Henze, G.; et al. Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF. Leukemia 2004, 18, 926–933. [Google Scholar] [CrossRef]
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- He, Y.; Wang, Y.; Liu, H.; Xu, X.; He, S.; Tang, J.; Huang, Y.; Miao, X.; Wu, Y.; Wang, Q.; et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leuk. Res. 2015, 39, 1428–1436. [Google Scholar] [CrossRef]
- Ikeda, S.; Abe, F.; Matsuda, Y.; Kitadate, A.; Takahashi, N.; Tagawa, H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci. 2020, 111, 4088–4101. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, X.; Feng, J.; Zhang, X. Overexpression of miR-202 resensitizes imatinib resistant chronic myeloid leukemia cells through targetting Hexokinase 2. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-F.; Jiang, S.; Liu, M.-F. MicroRNA regulation and analytical methods in cancer cell metabolism. Experientia 2017, 144, 646–2941. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Cheng, Z.; Pang, Y.; Zhao, H.; Chen, L.; Wang, C.; Qin, T.; Li, Q.; Han, Y.; Shi, J.; et al. High expression of MiR-98 is a good prognostic factor in acute myeloid leukemia patients treated with chemotherapy alone. J. Cancer 2019, 10, 178–185. [Google Scholar] [CrossRef]
- Xia, L.; Jiang, Y.; Zhang, X.-H.; Wang, X.-R.; Wei, R.; Qin, K.; Lu, Y. SUMOylation disassembles the tetrameric pyruvate kinase M2 to block myeloid differentiation of leukemia cells. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose Metabolism as a Target of Histone Deacetylase Inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef]
- McDonnell, S.R.P.; Hwang, S.; Rolland, D.; Murga-Zamalloa, C.; Basrur, V.; Conlon, K.P.; Fermin, D.; Wolfe, T.; Raskind, A.; Ruan, C.; et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK–mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood 2013, 122, 958–968. [Google Scholar] [CrossRef]
- Nie, Z.-Y.; Liu, X.-J.; Zhan, Y.; Liu, M.-H.; Zhang, X.-Y.; Li, Z.-Y.; Lu, Y.-Q.; Luo, J.-M.; Yang, L. miR-140-5p induces cell apoptosis and decreases Warburg effect in chronic myeloid leukemia by targeting SIX1. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [CrossRef]
- Agirre, X.; Vilas-Zornoza, A.; Jiménez-Velasco, A.; Martin-Subero, J.I.; Cordeu, L.; Garate, L.; José-Eneriz, E.S.; Abizanda, G.; Rodriguez-Otero, P.; Fortes, P.; et al. Epigenetic Silencing of the Tumor Suppressor MicroRNA Hsa-miR-124a Regulates CDK6 Expression and Confers a Poor Prognosis in Acute Lymphoblastic Leukemia. Cancer Res. 2009, 69, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; So, C.C.; Loong, F.; Chung, L.P.; Lam, W.W.L.; Liang, R.; Li, G.K.H.; Jin, N.-Y.; Chim, C. Epigenetic Inactivation of the miR-124-1 in Haematological Malignancies. PLoS ONE 2011, 6, e19027. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Y.; Li, H.H.; Zhu, X.C.; Dong, Y.W.; Fu, D.; Zhao, Q.L.; Wu, W.; Wu, X.Z. MiR-223 Suppresses Cell Proliferation by Targeting IGF-1R. PLoS ONE 2011, 6, e27008. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; Amodio, N.; DI Martino, M.T.; Altomare, E.; Leotta, M.; Caracciolo, D.; Gullà, A.; Neri, A.; Taverna, S.; D’Aquila, P.; et al. Targeting of multiple myeloma-related angiogenesis by miR-199a-5p mimics: In vitro and in vivo anti-tumor activity. Oncotarget 2014, 5, 3039–3054. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, H.-Y.; Zhang, J.-L.; Wan, D.-M.; Li, Y.-M.; Jiang, Z.-X. Dysregulation of LncRNA ANRIL mediated by miR-411–3p inhibits the malignant proliferation and tumor stem cell like property of multiple myeloma via hypoxia-inducible factor 1α. Exp. Cell Res. 2020, 396, 112280. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shanafelt, T.D.; Cimmino, A.; Taccioli, C.; Volinia, S.; Liu, C.-G.; Calin, G.A.; Croce, C.M.; Chan, D.A.; Giaccia, A.J.; et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood 2009, 113, 5568–5574. [Google Scholar] [CrossRef]
- Takeuchi, A.; Nishioka, C.; Ikezoe, T.; Yang, J.; Yokoyama, A. STAT5A regulates DNMT3A in CD34+/CD38− AML cells. Leuk. Res. 2015, 39, 897–905. [Google Scholar] [CrossRef]
- Luo, J.; Mu, J.; Zhang, M.; Zhao, B.; Liu, L. SPAG6-silencing enhances decitabine-induced apoptosis and demethylation of PTEN in SKM-1 cells and in a xenograft mouse model. Leuk. Lymphoma 2021, 1–12. [Google Scholar] [CrossRef]
- Piras, G.; Monne, M.; Palmas, A.D.; Calvisi, A.; Asproni, R.; Vacca, F.; Pilo, L.; Gabbas, A.; Latte, G. Methylation analysis of the phosphates and tensin homologue on chromosome 10 gene (PTEN) in multiple myeloma. Clin. Epigenetics 2014, 6, 16. [Google Scholar] [CrossRef]
- Kanduri, M.; Cahill, N.; Göransson, H.; Enström, C.; Ryan, F.; Isaksson, A.; Rosenquist, R. Differential genome-wide array–based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010, 115, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Labno-Kirszniok, K.; Nieszporek, T.; Wiecek, A.; Helbig, G.; Lubinski, J. Acute myeloid leukemia in a 38-year-old hemodialyzed patient with von Hippel-Lindau disease. Hered. Cancer Clin. Pract. 2013, 11, 11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Place, T.L.; Fitzgerald, M.P.; Venkataraman, S.; Vorrink, S.U.; Case, A.J.; Teoh, M.L.T.; Domann, F.E. Aberrant Promoter CpG Methylation Is a Mechanism for Impaired PHD3 Expression in a Diverse Set of Malignant Cells. PLoS ONE 2011, 6, e14617. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ohguchi, H.; Hideshima, T.; Bhasin, M.; Gorgun, G.T.; Santo, L.; Cea, M.; Samur, M.K.; Mimura, N.; Suzuki, R.; Tai, Y.-T.; et al. The KDM3A–KLF2–IRF4 axis maintains myeloma cell survival. Nat. Commun. 2016, 7, 10258. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell. Physiol. Biochem. 2017, 41, 921–932. [Google Scholar] [CrossRef]
- Liu, M.; Gao, J.; Huang, Q.; Jin, Y.; Wei, Z. Downregulating microRNA-144 mediates a metabolic shift in lung cancer cells by regulating GLUT1 expression. Oncol. Lett. 2016, 11, 3772–3776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, J.; Chen, S.; Shen, K.; Ai, G.; Dai, X.; Xie, B.; Shi, Y.; Jiang, S.; Feng, J.; et al. Decreased miR-144 expression as a non-invasive biomarker for acute myeloid leukemia patients. Die Pharm. 2017, 72, 232–235. [Google Scholar]
- Gupta, N.; Kumar, R.; Sharma, A. Inhibition of miR-144/199 promote myeloma pathogenesis via upregulation of versican and FAK/STAT3 signaling. Mol. Cell. Biochem. 2021, 476, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, Z.; Lin, J.; Liu, P. MiR-144-3p inhibits cell proliferation and induces apoptosis in multiple myeloma by targeting c-Met. Am. J. Transl. Res. 2017, 9, 2437–2446. [Google Scholar]
- Chen, H.; Cao, W.; Chen, J.; Liu, D.; Zhou, L.; Du, F.; Zhu, F. miR-218 contributes to drug resistance in multiple myeloma via targeting LRRC28. J. Cell. Biochem. 2021, 122, 305–314. [Google Scholar] [CrossRef]
- Jiang, L.; Meng, W.; Yu, G.; Yin, C.; Wang, Z.; Liao, L.; Meng, F. MicroRNA-144 targets APP to regulate AML1/ETO+ leukemia cell migration via the p-ERK/c-Myc/MMP-2 pathway. Oncol. Lett. 2019, 18, 2034–2042. [Google Scholar] [CrossRef]
- Li, Z.; Wong, K.Y.; Calin, G.A.; Chng, W.-J.; Chan, G.C.-F.; Chim, C.S. Epigenetic silencing of miR-340-5p in multiple myeloma: Mechanisms and prognostic impact. Clin. Epigenetics 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Wang, Q.; Feng, T.; Xu, J.; Miao, M.; Ji, X.; Zhu, H.; Shao, X. Low expression of microRNA-340 confers adverse clinical outcome in patients with acute myeloid leukemia. J. Cell. Physiol. 2018, 234, 4200–4205. [Google Scholar] [CrossRef]
- King, B.; Esguerra, J.L.S.; Golec, E.; Eliasson, L.; Kemper, C.; Blom, A.M. CD46 Activation Regulates miR-150–Mediated Control of GLUT1 Expression and Cytokine Secretion in Human CD4+ T Cells. J. Immunol. 2016, 196, 1636–1645. [Google Scholar] [CrossRef]
- Srutova, K.; Curik, N.; Burda, P.; Savvulidi, F.G.; Silvestri, G.; Trotta, R.; Klamova, H.; Pecherkova, P.; Sovova, Z.; Koblihova, J.; et al. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica 2018, 103, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, J. miR-150: Targeting MLL leukemia. Oncotarget 2012, 3, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Q.; Dai, Y.; Jiang, T.; Zhou, Y. miR-532-3p Inhibits Proliferation and Promotes Apoptosis of Lymphoma Cells by Targeting β-Catenin. J. Cancer 2020, 11, 4762–4770. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Smith, K.R.; Khong, H.T.; Huang, J.; Ahn, E.-Y.E.; Zhou, M.; Tan, M. miR-125b regulates differentiation and metabolic reprogramming of T cell acute lymphoblastic leukemia by directly targeting A20. Oncotarget 2016, 7, 78667–78679. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tili, E.; Michaille, J.-J.; Luo, Z.; Volinia, S.; Rassenti, L.Z.; Kipps, T.J.; Croce, C.M. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood 2012, 120, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.-F.F.; Mankaruos, M.; Scorilas, A.; Youssef, Y.; Girgis, A.; Mossad, S.; Metias, S.; Rofael, Y.; Honey, R.J.; Stewart, R.; et al. The miR-17-92 Cluster is Over Expressed in and Has an Oncogenic Effect on Renal Cell Carcinoma. J. Urol. 2010, 183, 743–751. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, Y.; Chen, S.; Zheng, C.; Hu, J.; Luo, S. MiR-19a regulates the cell growth and apoptosis of osteosarcoma stem cells by targeting PTEN. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Hao, M.; Zang, M.; Wendlandt, E.; Xu, Y.; An, G.; Gong, D.; Li, F.; Qi, F.; Zhang, Y.; Yang, Y.; et al. Low serum miR-19a expression as a novel poor prognostic indicator in multiple myeloma. Int. J. Cancer 2014, 136, 1835–1844. [Google Scholar] [CrossRef]
- Cao, L.; Wang, N.; Pan, J.; Hu, S.; Zhao, W.; He, H.; Wang, Y.; Gu, G.; Chai, Y. Clinical significance of microRNA-34b expression in pediatric acute leukemia. Mol. Med. Rep. 2016, 13, 2777–2784. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; Wu, T.; Yang, S. miR-489 suppresses multiple myeloma cells growth through inhibition of LDHA-mediated aerobic glycolysis. Genes Genom. 2019, 42, 291–297. [Google Scholar] [CrossRef]
- Qi, H.-X.; Cao, Q.; Zhou, G.-P.; Sun, X.-Z.; Zhou, W.-D.; Hong, Z.; Hu, J.; Juan, C.-X.; Li, S.; Kuai, W.-X. MicroRNA 34b inhibits cell proliferation in pediatric acute myeloid leukemia via regulating LDHA. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5351–5359. [Google Scholar]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar] [CrossRef]
- Kohrogi, K.; Hino, S.; Sakamoto, A.; Anan, K.; Takase, R.; Araki, H.; Hino, Y.; Araki, K.; Sato, T.; Nakamura, K.; et al. LSD1 defines erythroleukemia metabolism by controlling the lineage-specific transcription factors GATA1 and C/EBPα. Blood Adv. 2021, 5, 2305–2318. [Google Scholar] [CrossRef]
- Madan, V.; Koeffler, H.P. Differentiation therapy of myeloid leukemia: Four decades of development. Haematologica 2020, 106, 1–13. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Powers, J.T.; Sacco, A.; Glavey, S.V.; Huynh, D.; Reagan, M.R.; Salem, K.Z.; Moschetta, M.; Shi, J.; Mishima, Y.; et al. The LIN28B/let-7 axis is a novel therapeutic pathway in multiple myeloma. Leukemia 2016, 31, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Ching, Y.Q.; Chooi, J.-Y.; Lu, X.; Quah, J.Y.; Toh, S.H.-M.; Chan, Z.-L.; Tan, T.Z.; Chong, P.S.; et al. Inhibition of LIN28B impairs leukemia cell growth and metabolism in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Chen, B.; Liu, Y.; Jin, X.; Lu, W.; Liu, J.; Xia, Z.; Yuan, Q.; Zhao, X.; Xu, N.; Liang, S. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer 2014, 14, 443. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, G.; Yan, W.; Zhan, H.; Sun, P. MiR-146b-5p Regulates Cell Growth, Invasion, and Metabolism by Targeting PDHB in Colorectal Cancer. Am. J. Cancer Res. 2017, 7, 1136. [Google Scholar]
- Wei, S.; Ma, W. MiR-370 functions as oncogene in melanoma by direct targeting pyruvate dehydrogenase B. Biomed. Pharmacother. 2017, 90, 278–286. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.; Fang, C.; Li, C.; Xhyliu, F.; Dysert, H.; Bodo, J.; Habermehl, G.; Russell, B.E.; Li, W.; et al. Targeting of CD38 by the tumor suppressor miR-26a serves as a novel potential therapeutic agent in multiple myeloma. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Xiong, J.; Xiao, R.; Shao, L.; Yang, X.; Zhou, L.; Yuan, W.; Wang, M.; Yin, Q.; Wu, Y.; et al. Loss of miR-146b-5p promotes T cell acute lymphoblastic leukemia migration and invasion via the IL-17A pathway. J. Cell. Biochem. 2018, 120, 5936–5948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, J.; Zhou, M.; Li, B.; Zhang, Y.; Huang, T.; Wang, L.; Jia, J.; Chen, C. The tumor suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid leukemia. Mol. Cancer 2012, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- D’Abundo, L.; Callegari, E.; Bresin, A.; Chillemi, A.; Elamin, B.K.; Guerriero, P.; Huang, X.; Saccenti, E.; Hussein, E.M.; Casciano, F.; et al. Anti-leukemic activity of microRNA-26a in a chronic lymphocytic leukemia mouse model. Oncogene 2017, 36, 6617–6626. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xiao, D.; Zhao, Z.; Ren, P.; Li, C.; Hu, Y.; Shi, J.; Su, H.; Wang, L.; Liu, H.; et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis 2017, 6, e356. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, S.; Deng, S.; An, G.; Qin, X.; Li, F.; Xu, Y.; Hao, M.; Yang, Y.; Zhou, W.; et al. Epigenetic silencing of miR-137 induces drug resistance and chromosomal instability by targeting AURKA in multiple myeloma. Leukemia 2016, 31, 1123–1135. [Google Scholar] [CrossRef]
- Huang, Y.; Zou, Y.; Zheng, R.; Ma, X. MiR-137 inhibits cell proliferation in acute lymphoblastic leukemia by targeting JARID1B. Eur. J. Haematol. 2019, 103, 215–224. [Google Scholar] [CrossRef]
- Zhang, T.-J.; Qian, Z.; Wen, X.-M.; Zhou, J.-D.; Li, X.-X.; Xu, Z.-J.; Ma, J.-C.; Zhang, Z.-H.; Lin, J.; Qian, J. Lower expression of bone marrow miR-122 is an independent risk factor for overall survival in cytogenetically normal acute myeloid leukemia. Pathol. Res. Pr. 2018, 214, 896–901. [Google Scholar] [CrossRef]
- Wang, N.; Tan, H.-Y.; Feng, Y.-G.; Zhang, C.; Chen, F.; Feng, Y. microRNA-23a in Human Cancer: Its Roles, Mechanisms and Therapeutic Relevance. Cancers 2018, 11, 7. [Google Scholar] [CrossRef]
- Jeong, S.M.; Haigis, A.M.C. Sirtuins in Cancer: A Balancing Act between Genome Stability and Metabolism. Mol. Cells 2015, 38, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Li, X.; Kazgan, N. Mammalian Sirtuins and Energy Metabolism. Int. J. Biol. Sci. 2011, 7, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.; Camporez, J.P.; Ruan, H.-B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The Deacetylase Sirt6 Activates the Acetyltransferase GCN5 and Suppresses Hepatic Gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Basappa, J.; Citir, M.; Zhang, Q.; Wang, H.Y.; Liu, X.; Melnikov, O.; Yahya, H.; Stein, F.; Muller, R.; Traynor-Kaplan, A.; et al. ACLY is the novel signaling target of PIP2/PIP3 and Lyn in acute myeloid leukemia. Heliyon 2020, 6, e03910. [Google Scholar] [CrossRef]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.-P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef]
- Trissal, M.C.; Wong, T.N.; Yao, J.-C.; Ramaswamy, R.; Kuo, I.; Baty, J.D.; Sun, Y.; Jih, G.; Parikh, N.; Berrien-Elliott, M.M.; et al. MIR142 loss-of-function mutations derepress ASH1L to increase HOXA gene expression and promote leukemogenesis. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of -KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.; White, D.A.; Drayson, M.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.-H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 Has Tumor-Suppressive Activity and Regulates the Cellular Metabolic Response to DNA Damage by Inhibiting Mitochondrial Glutamine Metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Wei, W.; Sun, W.; Yu, S.; Yang, Y.; Ai, L. Butyrate production from high-fiber diet protects against lymphoma tumor. Leuk. Lymphoma 2016, 57, 2401–2408. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- Momparler, R.L.; Côté, S.; Momparler, L.F.; Idaghdour, Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin. Epigenetics 2014, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.-K.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-β-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Cerone, R.; Schiaffino, M.C.; Fantasia, A.R.; Minniti, G.; Caruso, U.; Lorini, R.; Watkins, D.; Matiaszuk, N.; Rosenblatt, D.S.; et al. GlycineN-methyltransferase deficiency: A novel inborn error causing persistent isolated hypermethioninaemia. J. Inherit. Metab. Dis. 2001, 24, 448–464. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nature 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- de Jonge, R.; Tissing, W.J.E.; Hooijberg, J.H.; Jansen, G.; Kaspers, G.J.L.; Lindemans, J.; Peters, G.J.; Pieters, R. Polymorphisms in folate-related genes and risk of pediatric acute lymphoblastic leukemia. Blood 2009, 113, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- Nylund, P.; Párraga, A.A.; Haglöf, J.; De Bruyne, E.; Menu, E.; Garrido-Zabala, B.; Ma, A.; Jin, J.; Öberg, F.; Vanderkerken, K.; et al. A distinct metabolic response characterizes sensitivity to EZH2 inhibition in multiple myeloma. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Salati, S.; Salvestrini, V.; Carretta, C.; Genovese, E.; Rontauroli, S.; Zini, R.; Rossi, C.; Ruberti, S.; Bianchi, E.; Barbieri, G.; et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget 2017, 8, 49451–49469. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zheng, G.; Zhuang, H.; Li, X.; Hu, D.; Zhu, L.; Wang, T.; You, M.J.; Zhang, Y. MicroRNA-494 Activation Suppresses Bone Marrow Stromal Cell-Mediated Drug Resistance in Acute Myeloid Leukemia Cells. J. Cell. Physiol. 2016, 232, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, Y.; Chen, Z.; Shi, Y.; He, Z.; Ding, B.; Wang, C.; Yu, L. miR-134 increases the antitumor effects of cytarabine by targeting Mnks in acute myeloid leukemia cells. OncoTargets Ther. 2018, ume 11, 3141–3147. [Google Scholar] [CrossRef]
- Tian, C.; Zhang, L.; Li, X.; Zhang, Y.; Li, J.; Chen, L. Low miR-192 expression predicts poor prognosis in pediatric acute myeloid leukemia. Cancer Biomark. 2018, 22, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pan, J.; Mao, S.; Jin, J. IL-17/miR-192/IL-17Rs Regulatory Feedback Loop Facilitates Multiple Myeloma Progression. PLoS ONE 2014, 9, e114647. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Guo, H.; Bates, P.D.; Zhang, S.; Zhang, H.; Nomie, K.J.; Li, Y.; Lu, L.; Seibold, K.R.; Wang, F.; et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019, 33, 2898–2911. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Batova, A.; Shao, L.; Carrera, C.J.; Yu, A. Presence of methylthioadenosine phosphorylase (MTAP) in hematopoietic stem/progenitor cells: Its therapeutic implication for MTAP (-) malignancies. Clin. Cancer Res. 1997, 3, 433–438. [Google Scholar] [PubMed]
- Nepali, K.; Liou, J.-P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 1–58. [Google Scholar] [CrossRef]
- Danenberg, P.V.; Gustavsson, B.; Johnston, P.; Lindberg, P.; Moser, R.; Odin, E.; Peters, G.J.; Petrelli, N. Folates as adjuvants to anticancer agents: Chemical rationale and mechanism of action. Crit. Rev. Oncol. 2016, 106, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.; Nordström, A. Current Status and Future Prospects of Clinically Exploiting Cancer-specific Metabolism-Why Is Tumor Metabolism Not More Extensively Translated into Clinical Targets and Biomarkers? Int. J. Mol. Sci. 2019, 20, 1385. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalushkova, A.; Nylund, P.; Párraga, A.A.; Lennartsson, A.; Jernberg-Wiklund, H. One Omics Approach Does Not Rule Them All: The Metabolome and the Epigenome Join Forces in Haematological Malignancies. Epigenomes 2021, 5, 22. https://doi.org/10.3390/epigenomes5040022
Kalushkova A, Nylund P, Párraga AA, Lennartsson A, Jernberg-Wiklund H. One Omics Approach Does Not Rule Them All: The Metabolome and the Epigenome Join Forces in Haematological Malignancies. Epigenomes. 2021; 5(4):22. https://doi.org/10.3390/epigenomes5040022
Chicago/Turabian StyleKalushkova, Antonia, Patrick Nylund, Alba Atienza Párraga, Andreas Lennartsson, and Helena Jernberg-Wiklund. 2021. "One Omics Approach Does Not Rule Them All: The Metabolome and the Epigenome Join Forces in Haematological Malignancies" Epigenomes 5, no. 4: 22. https://doi.org/10.3390/epigenomes5040022
APA StyleKalushkova, A., Nylund, P., Párraga, A. A., Lennartsson, A., & Jernberg-Wiklund, H. (2021). One Omics Approach Does Not Rule Them All: The Metabolome and the Epigenome Join Forces in Haematological Malignancies. Epigenomes, 5(4), 22. https://doi.org/10.3390/epigenomes5040022