
Evolution of CG Methylation Maintenance Machinery in Plants


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
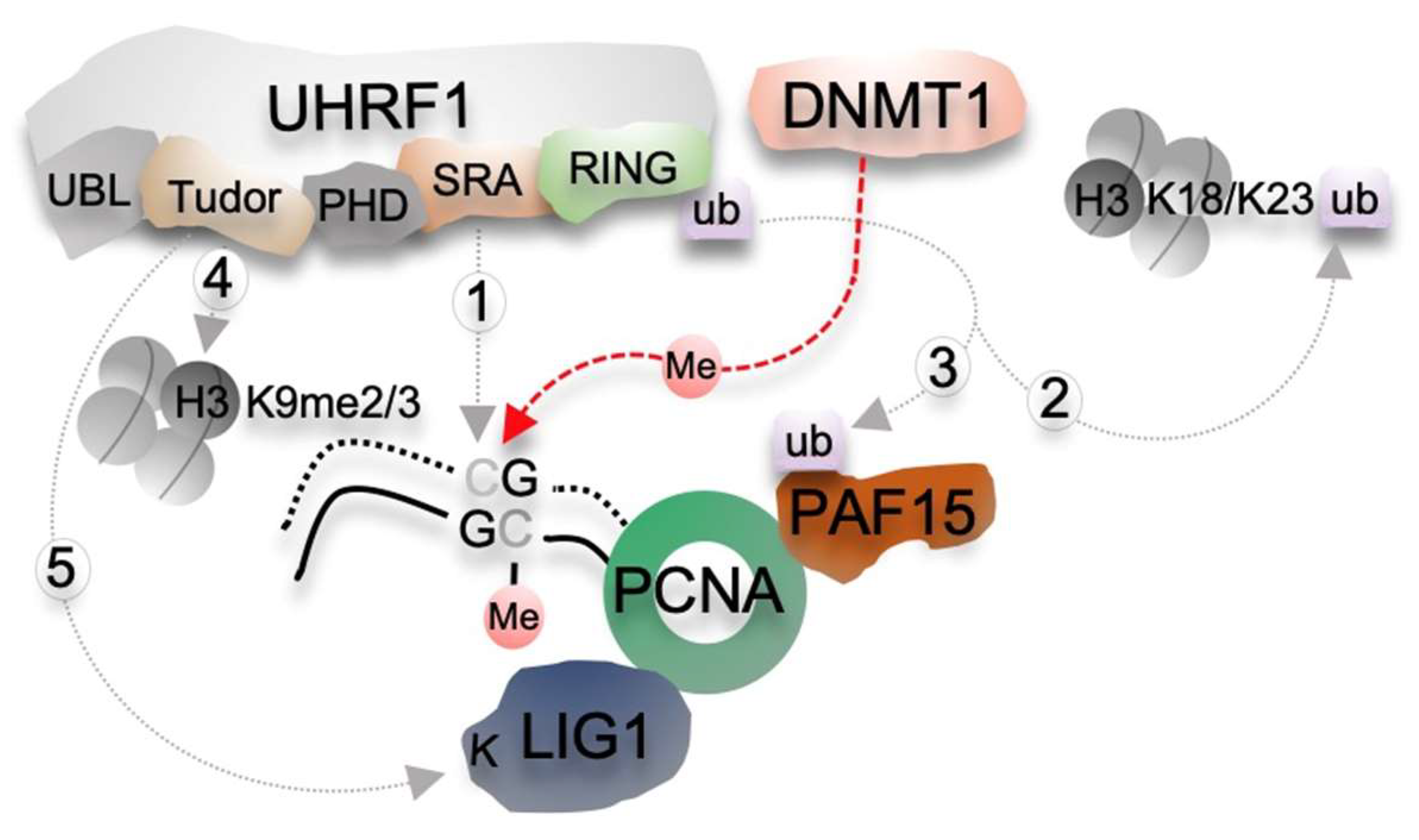
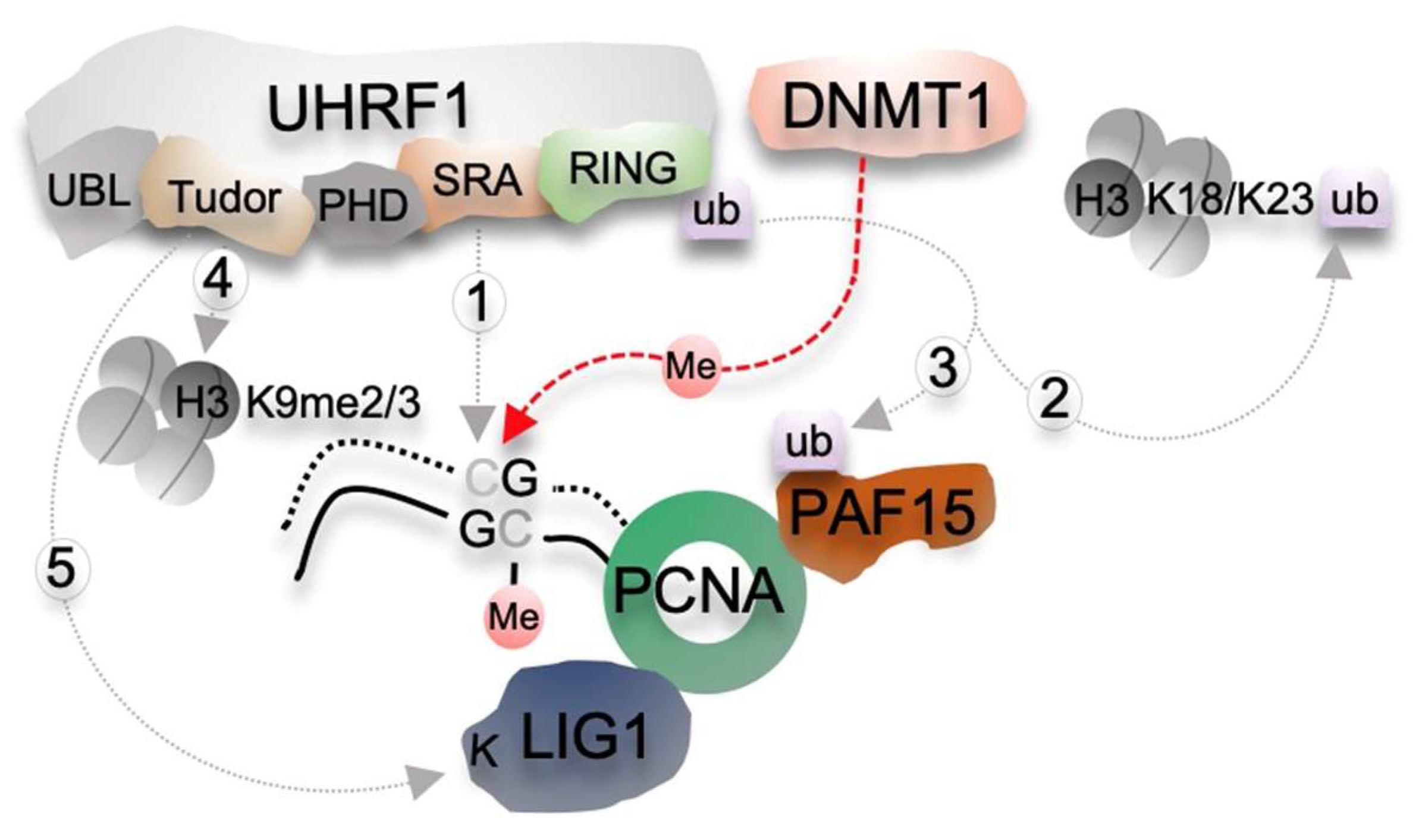
2. Molecular Mechanisms of the DNMT1/UHRF1 Pathway
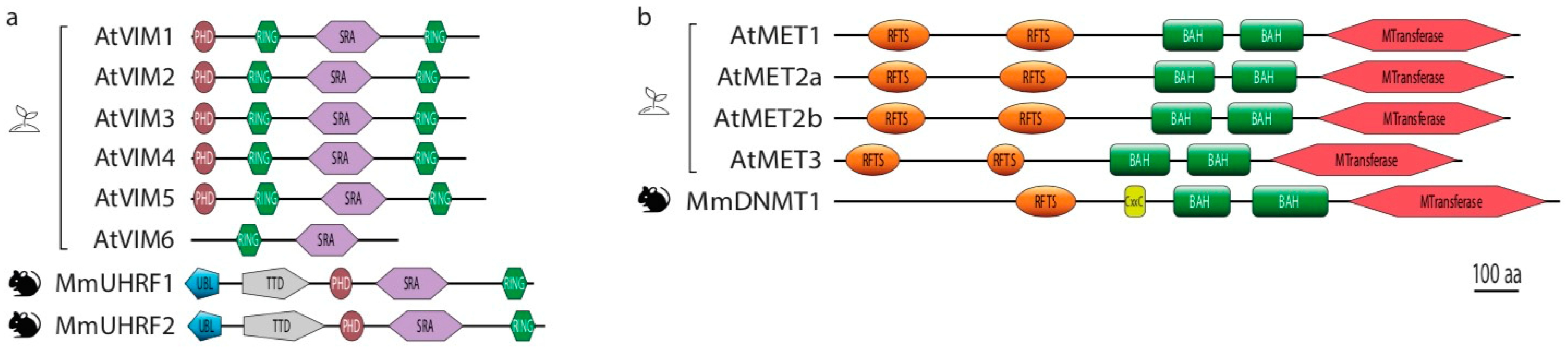
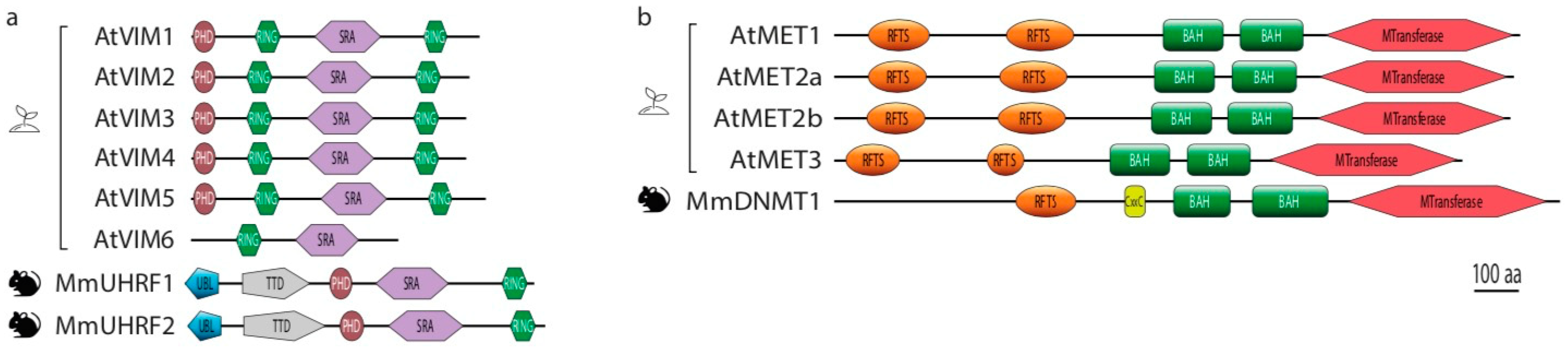
3. Molecular Mechanisms of the MET/VIM Pathway in Plants
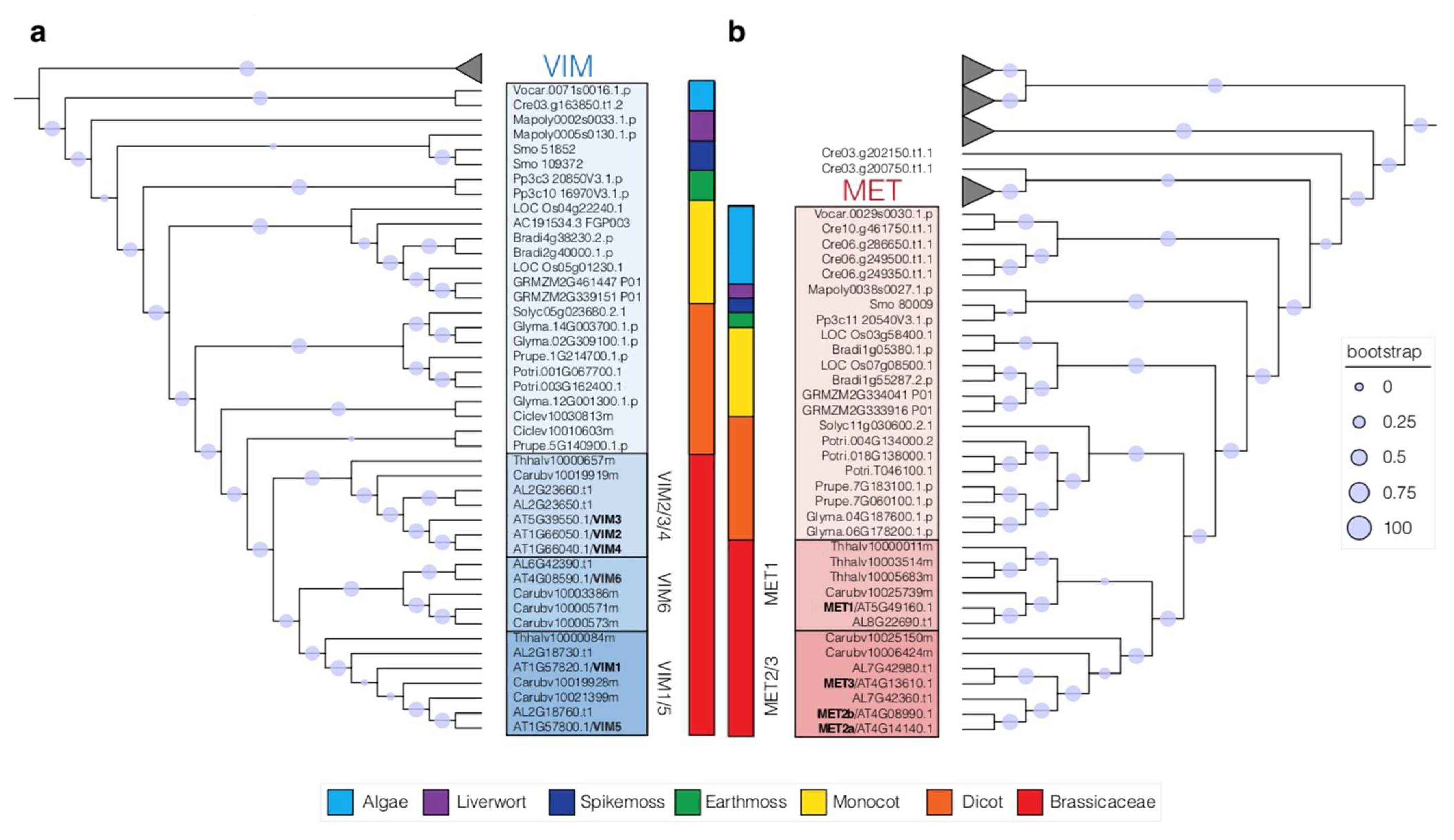
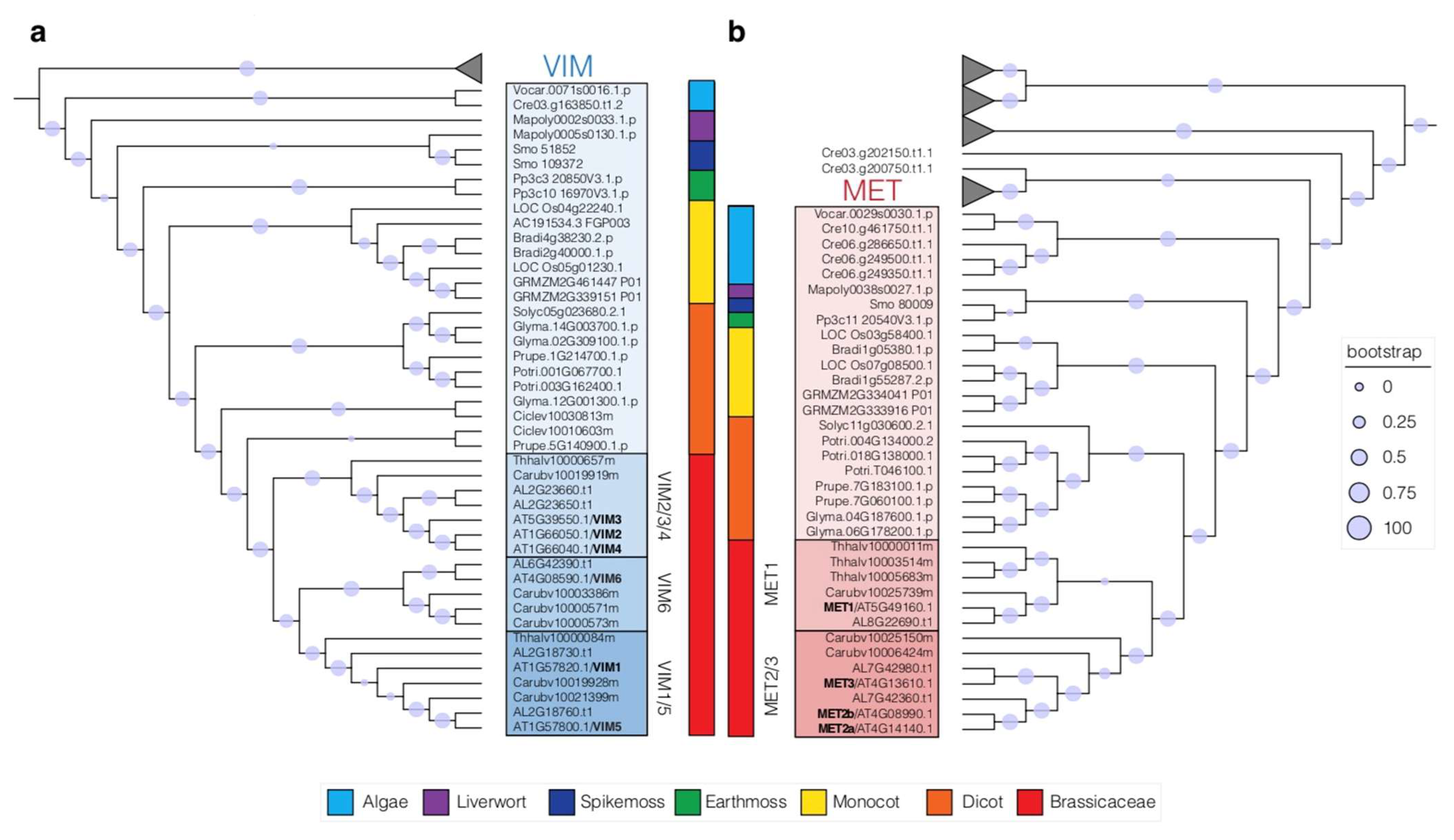
4. Duplication of the MET and VIM Proteins in Plants
5. Alternative MET/VIM Pathways during Reproduction
6. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Zilberman, D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 2010, 20, R780–R785. [Google Scholar] [CrossRef] [Green Version]
- Raddatz, G.; Guzzardo, P.M.; Olova, N.; Fantappie, M.R.; Rampp, M.; Schaefer, M.; Reik, W.; Hannon, G.J.; Lyko, F. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc. Natl. Acad. Sci. USA 2013, 110, 8627–8631. [Google Scholar] [CrossRef] [Green Version]
- Feil, R.; Berger, F. Convergent evolution of genomic imprinting in plants and mammals. Trends Genet. 2007, 23, 192–199. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. X-Chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annu. Rev. Genet. 2018, 52, 535–566. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Brautigam, K.; Cronk, Q. DNA Methylation and the evolution of developmental complexity in Plants. Front. Plant Sci. 2018, 9, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Fan, H.; Grimm, S.A.; Guo, Y.; Kim, J.J.; Yin, J.; Li, L.; Petell, C.J.; Tan, X.F.; Zhang, Z.M.; et al. Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc. Natl. Acad. Sci. USA 2020, 117, 18439–18447. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Matzke, M.A.; Kanno, T.; Matzke, A.J. RNA-Directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [Green Version]
- Ebbs, M.L.; Bender, J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.M.; Bostick, M.; Zhang, X.; Kraft, E.; Henderson, I.; Callis, J.; Jacobsen, S.E. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldan-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Kendall, T.; Forsythe, E.S.; Dorantes-Acosta, A.; Li, S.; Caballero-Perez, J.; Chen, X.; Arteaga-Vazquez, M.; Beilstein, M.A.; Mosher, R.A. Ancient origin and recent innovations of RNA Polymerase IV and V. Mol. Biol. Evol. 2015, 32, 1788–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xia, R.; Meyers, B.C.; Walbot, V. Evolution, functions, and mysteries of plant ARGONAUTE proteins. Curr. Opin. Plant Biol. 2015, 27, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.J.; Lewis, Z.A.; Goll, M.G. DNA Methylation: Shared and divergent features across eukaryotes. Trends Genet. 2019, 35, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Zhang, Z.; Zou, Z.; Lv, C.; Dong, Q.; He, Q.; Yi, Y.; Li, Y.; Wang, H.; Zhu, B. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res. 2020, 30, 980–996. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef]
- Petryk, N.; Bultmann, S.; Bartke, T.; Defossez, P.A. Staying true to yourself: Mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res. 2021, 49, 3020–3032. [Google Scholar] [CrossRef]
- Svedruzic, Z.M.; Reich, N.O. cytosine C5 methyltransferase Dnmt1: Catalysis-dependent release of allosteric inhibition. Biochemistry 2005, 44, 9472–9485. [Google Scholar] [CrossRef]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011, 331, 1036–1040. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, Q.; Li, P.; Liu, X.; Jia, Y.; Wu, W.; Li, J.; Dong, S.; Koseki, H.; Wong, J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011, 21, 1723–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowher, H.; Jeltsch, A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001, 309, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, F.; Fagan, R.L.; Wean, M.; Avvakumov, G.V.; Walker, J.R.; Xue, S.; Dhe-Paganon, S.; Brenner, C. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 2011, 286, 15344–15351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, K.; Suetake, I.; Yamashita, E.; Suga, M.; Narita, H.; Nakagawa, A.; Tajima, S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. USA 2011, 108, 9055–9059. [Google Scholar] [CrossRef] [Green Version]
- Schermelleh, L.; Haemmer, A.; Spada, F.; Rosing, N.; Meilinger, D.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007, 35, 4301–4312. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- Bashtrykov, P.; Jankevicius, G.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J. Biol. Chem. 2014, 289, 4106–4115. [Google Scholar] [CrossRef] [Green Version]
- Arita, K.; Isogai, S.; Oda, T.; Unoki, M.; Sugita, K.; Sekiyama, N.; Kuwata, K.; Hamamoto, R.; Tochio, H.; Sato, M.; et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. USA 2012, 109, 12950–12955. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Mulholland, C.B.; Bultmann, S.; Kori, S.; Endo, A.; Saeki, Y.; Qin, W.; Trummer, C.; Chiba, Y.; Yokoyama, H.; et al. Two distinct modes of DNMT1 recruitment ensure stable maintenance DNA methylation. Nat. Commun. 2020, 11, 1222. [Google Scholar] [CrossRef] [Green Version]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA Methylation. Mol. Cell 2017, 67, 550–565.e5. [Google Scholar] [CrossRef] [Green Version]
- Kori, S.; Ferry, L.; Matano, S.; Jimenji, T.; Kodera, N.; Tsusaka, T.; Matsumura, R.; Oda, T.; Sato, M.; Dohmae, N.; et al. Structure of the UHRF1 Tandem Tudor domain bound to a methylated non-histone protein, LIG1, reveals rules for binding and regulation. Structure 2019, 27, 485–496.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottach, A.; Frauer, C.; Pichler, G.; Bonapace, I.M.; Spada, F.; Leonhardt, H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010, 38, 1796–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nady, N.; Lemak, A.; Walker, J.R.; Avvakumov, G.V.; Kareta, M.S.; Achour, M.; Xue, S.; Duan, S.; Allali-Hassani, A.; Zuo, X.; et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011, 286, 24300–24311. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Pingault, L.; Poulet, A.; Duarte, J.; Throude, M.; Faure, S.; Pichon, J.P.; Paux, E.; Probst, A.V.; Tatout, C. Evolutionary history of Methyltransferase 1 genes in hexaploid wheat. BMC Genom. 2014, 15, 922. [Google Scholar] [CrossRef] [Green Version]
- Pei, L.; Zhang, L.; Li, J.; Shen, C.; Qiu, P.; Tu, L.; Zhang, X.; Wang, M. Tracing the origin and evolution history of methylation-related genes in plants. BMC Plant Biol. 2019, 19, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubko, E.; Gentry, M.; Kunova, A.; Meyer, P. De novo DNA methylation activity of methyltransferase 1 (MET1) partially restores body methylation in Arabidopsis thaliana. Plant J. 2012, 71, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Kraft, E.; Bostick, M.; Jacobsen, S.E.; Callis, J. ORTH/VIM proteins that regulate DNA methylation are functional ubiquitin E3 ligases. Plant J. 2008, 56, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Q.; Zhao, J.H.; Chen, Q.; Zhang, Z.H.; Li, J.; Guo, Z.X.; Xie, Q.; Ding, S.W.; Guo, H.S. DNA geminivirus infection induces an imprinted E3 ligase gene to epigenetically activate viral gene transcription. Plant Cell 2020, 32, 3256–3272. [Google Scholar] [CrossRef]
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008, 4, e1000156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunderland, P.A.; West, C.E.; Waterworth, W.M.; Bray, C.M. An evolutionarily conserved translation initiation mechanism regulates nuclear or mitochondrial targeting of DNA ligase 1 in Arabidopsis thaliana. Plant J. 2006, 47, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Ingouff, M.; Berger, F. Histone3 variants in plants. Chromosoma 2010, 119, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Pichler, G.; Wolf, P.; Schmidt, C.S.; Meilinger, D.; Schneider, K.; Frauer, C.; Fellinger, K.; Rottach, A.; Leonhardt, H. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell Biochem. 2011, 112, 2585–2593. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Xiong, J.; Wang, M.; Yang, N.; Wong, J.; Zhu, B.; Xu, R.M. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell 2014, 54, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Shook, M.S.; Richards, E.J. VIM proteins regulate transcription exclusively through the MET1 cytosine methylation pathway. Epigenetics 2014, 9, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Yaari, R.; Noy-Malka, C.; Wiedemann, G.; Auerbach Gershovitz, N.; Reski, R.; Katz, A.; Ohad, N. DNA METHYLTRANSFERASE 1 is involved in (m)CG and (m)CCG DNA methylation and is essential for sporophyte development in Physcomitrella patens. Plant Mol. Biol. 2015, 88, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Nishihama, R.; Yamaoka, S.; Arteaga-Vazquez, M.A.; Aguilar-Cruz, A.; Grimanelli, D.; Pogorelcnik, R.; Martienssen, R.A.; Yamato, K.T.; Kohchi, T.; et al. Loss of CG Methylation in Marchantia polymorpha Causes disorganization of cell division and reveals unique DNA Methylation regulatory mechanisms of non-CG methylation. Plant Cell Physiol. 2018, 59, 2421–2431. [Google Scholar] [CrossRef]
- Yamauchi, T.; Johzuka-Hisatomi, Y.; Terada, R.; Nakamura, I.; Iida, S. The MET1b gene encoding a maintenance DNA methyltransferase is indispensable for normal development in rice. Plant Mol. Biol. 2014, 85, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.R.; et al. Mutation of a major CG methylase in rice causes genomewide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef]
- Li, J.; Berger, F. Endosperm: Food for humankind and fodder for scientific discoveries. New Phytol. 2012, 195, 290–305. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A role for RNAi in the selective correction of DNA methylation defects. Science 2009, 323, 1600–1604. [Google Scholar] [CrossRef] [Green Version]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Muller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Schoft, V.K.; Chumak, N.; Mosiolek, M.; Slusarz, L.; Komnenovic, V.; Brownfield, L.; Twell, D.; Kakutani, T.; Tamaru, H. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 2009, 10, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijo, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef] [Green Version]
- Pignatta, D.; Erdmann, R.M.; Scheer, E.; Picard, C.L.; Bell, G.W.; Gehring, M. Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 2014, 3, e03198. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xia, H.; Zhang, Y.; Zhao, S.; Zhao, C.; Hou, L.; Li, C.; Li, A.; Ma, C.; Wang, X. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in Maize (Zea mays). BMC Genom. 2015, 16, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Park, K.; Kim, M.Y.; Vickers, M.; Park, J.S.; Hyun, Y.; Okamoto, T.; Zilberman, D.; Fischer, R.L.; Feng, X.; Choi, Y.; et al. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15138–15143. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Jullien, P.E.; Mosquna, A.; Ingouff, M.; Sakata, T.; Ohad, N.; Berger, F. Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol. 2008, 6, e194. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Susaki, D.; Yelagandula, R.; Higashiyama, T.; Berger, F. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 2012, 22, 1825–1830. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M. Genomic imprinting: Insights from plants. Annu. Rev. Genet. 2013, 47, 187–208. [Google Scholar] [CrossRef]
- Quadrana, L.; Bortolini Silveira, A.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. Elife 2016, 5, e15716. [Google Scholar] [CrossRef] [PubMed]
- Pagnussat, G.C.; Yu, H.J.; Ngo, Q.A.; Rajani, S.; Mayalagu, S.; Johnson, C.S.; Capron, A.; Xie, L.F.; Ye, D.; Sundaresan, V. Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development 2005, 132, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, R.; Sasaki, T.; Nishio, T. Characterization of DNA methyltransferase genes in Brassica rapa. Genes Genet. Syst. 2006, 81, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirot, L.; Jullien, P.E.; Ingouff, M. Evolution of CG Methylation Maintenance Machinery in Plants. Epigenomes 2021, 5, 19. https://doi.org/10.3390/epigenomes5030019
Tirot L, Jullien PE, Ingouff M. Evolution of CG Methylation Maintenance Machinery in Plants. Epigenomes. 2021; 5(3):19. https://doi.org/10.3390/epigenomes5030019
Chicago/Turabian StyleTirot, Louis, Pauline E. Jullien, and Mathieu Ingouff. 2021. "Evolution of CG Methylation Maintenance Machinery in Plants" Epigenomes 5, no. 3: 19. https://doi.org/10.3390/epigenomes5030019
APA StyleTirot, L., Jullien, P. E., & Ingouff, M. (2021). Evolution of CG Methylation Maintenance Machinery in Plants. Epigenomes, 5(3), 19. https://doi.org/10.3390/epigenomes5030019