
Helicase Lymphoid-Specific Enzyme Contributes to the Maintenance of Methylation of SST1 Pericentromeric Repeats That Are Frequently Demethylated in Colon Cancer and Associate with Genomic Damage

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
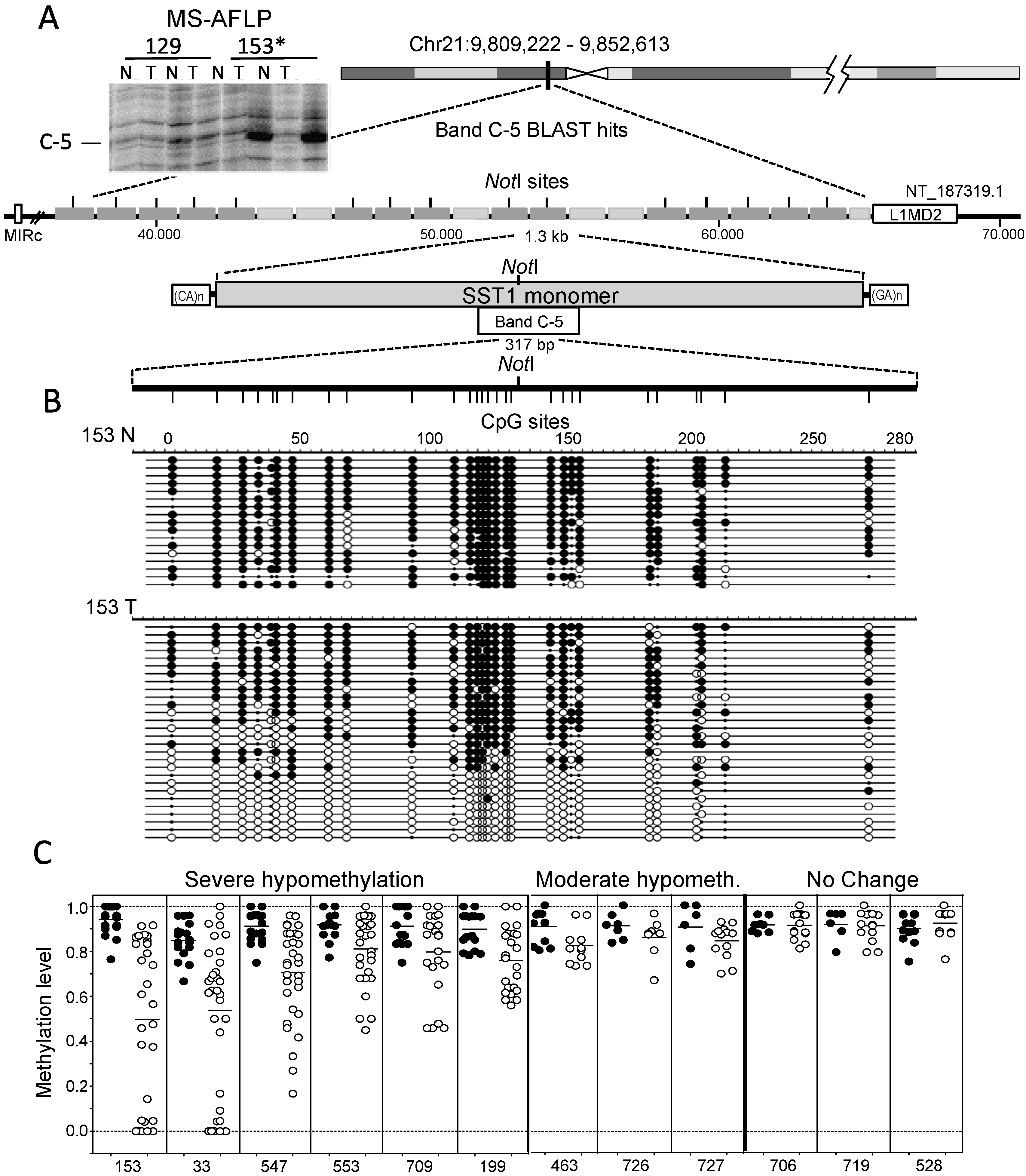
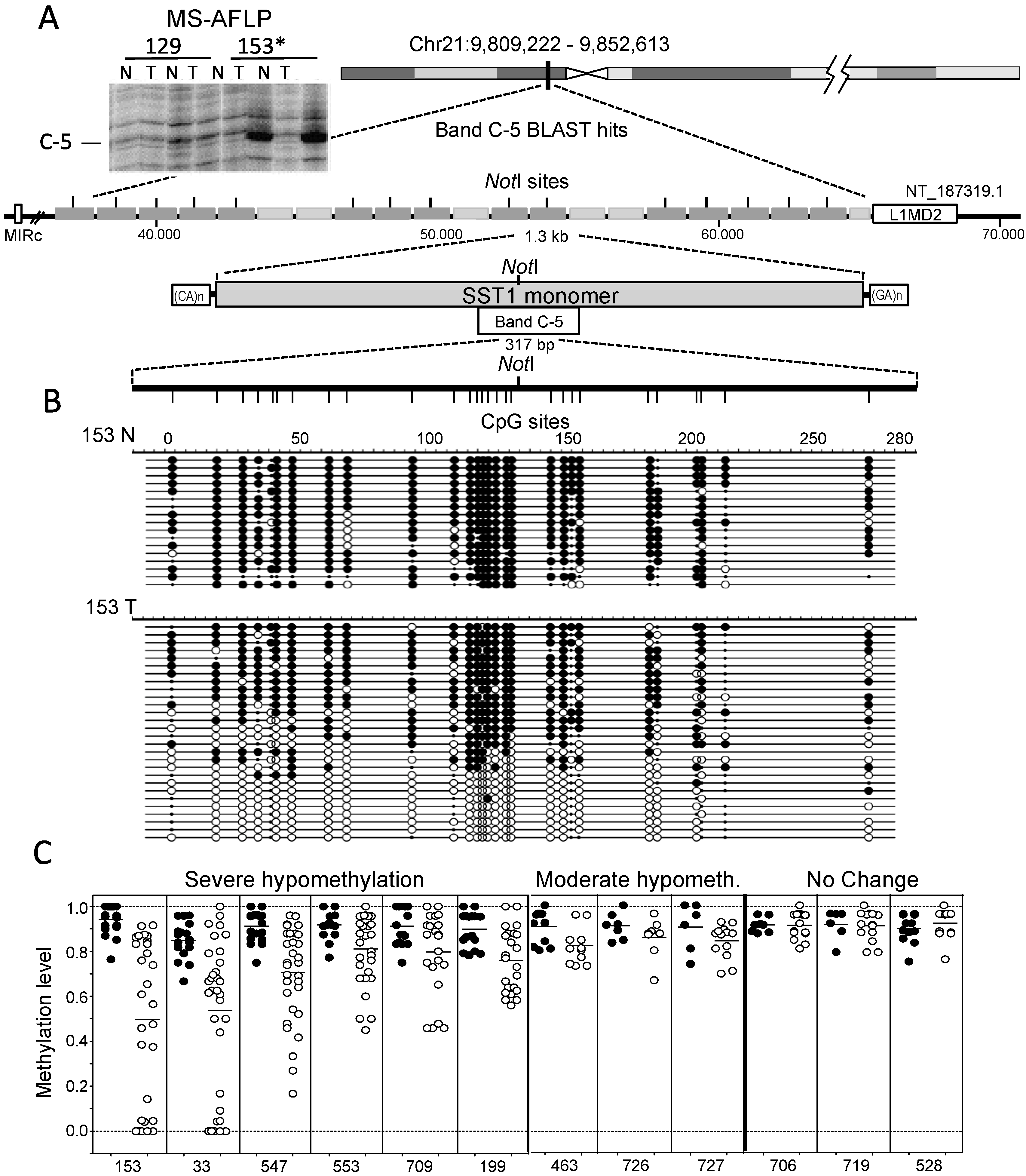
2.1. Demethylation of SST1 Pericentromeric Satellite Repeats in Human Cancers
2.2. SST1 Demethylation as a Marker of Global Hypomethylation
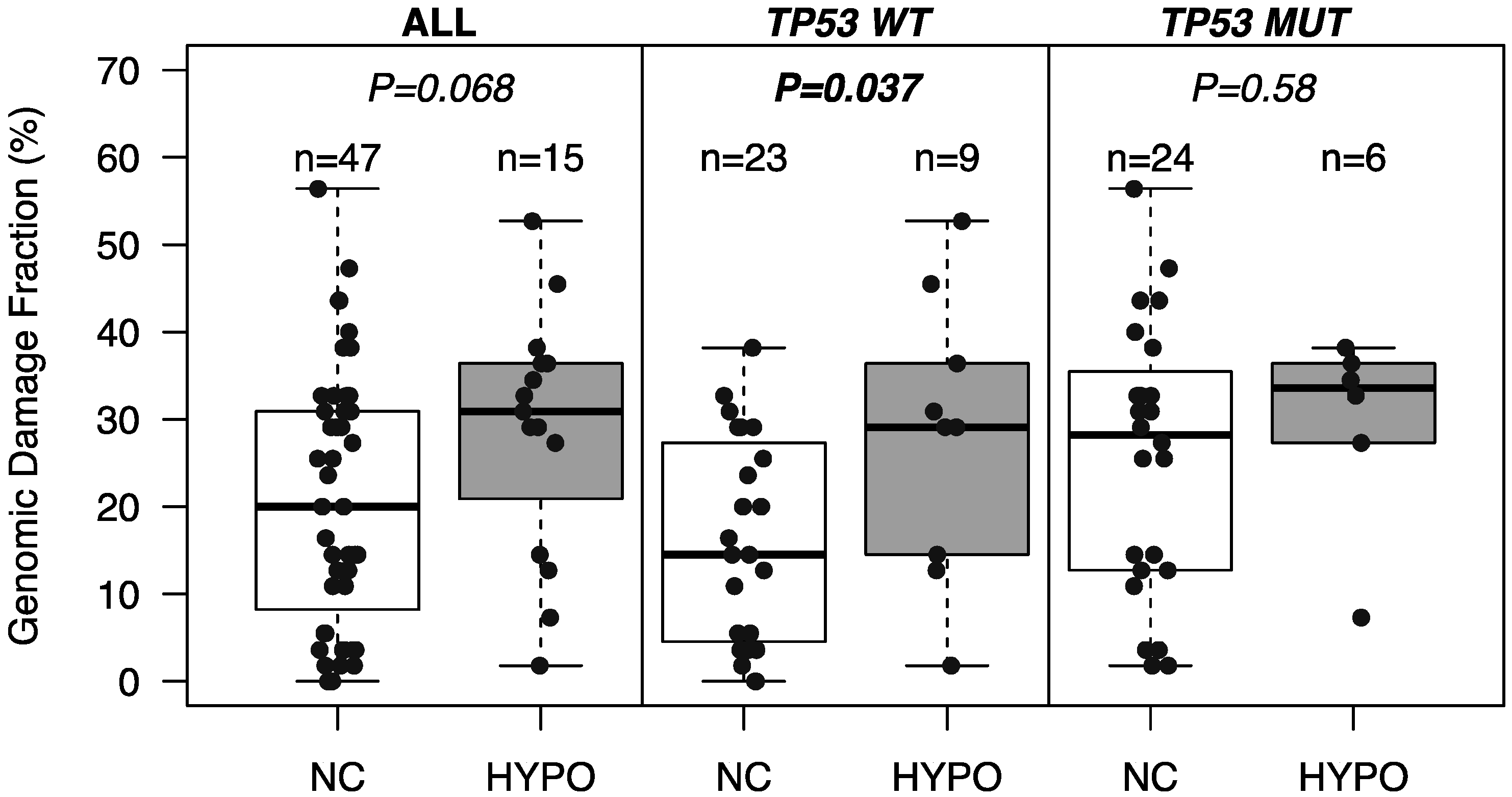
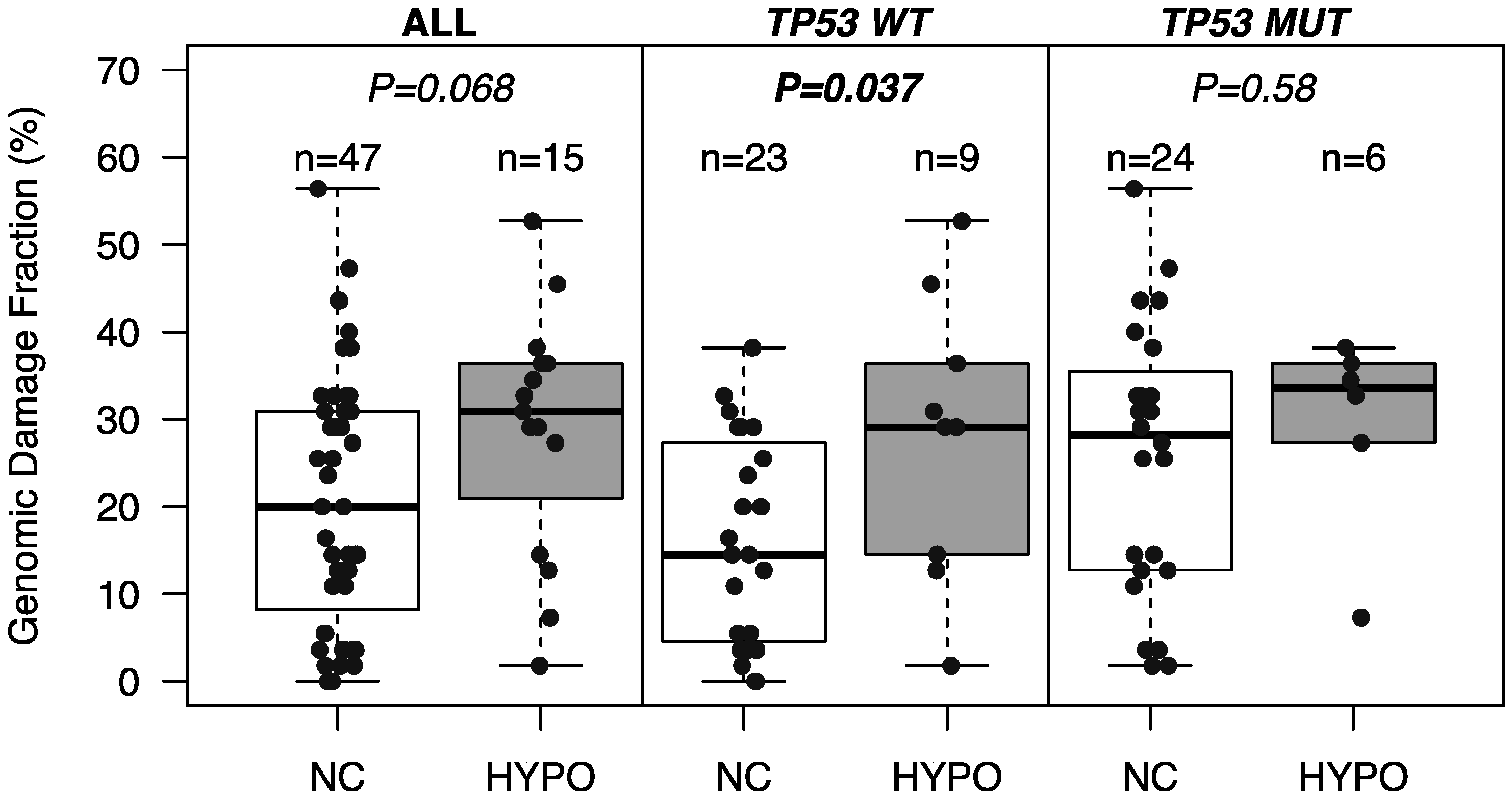
2.3. Association of SST1 Demethylation with Genomic Damage
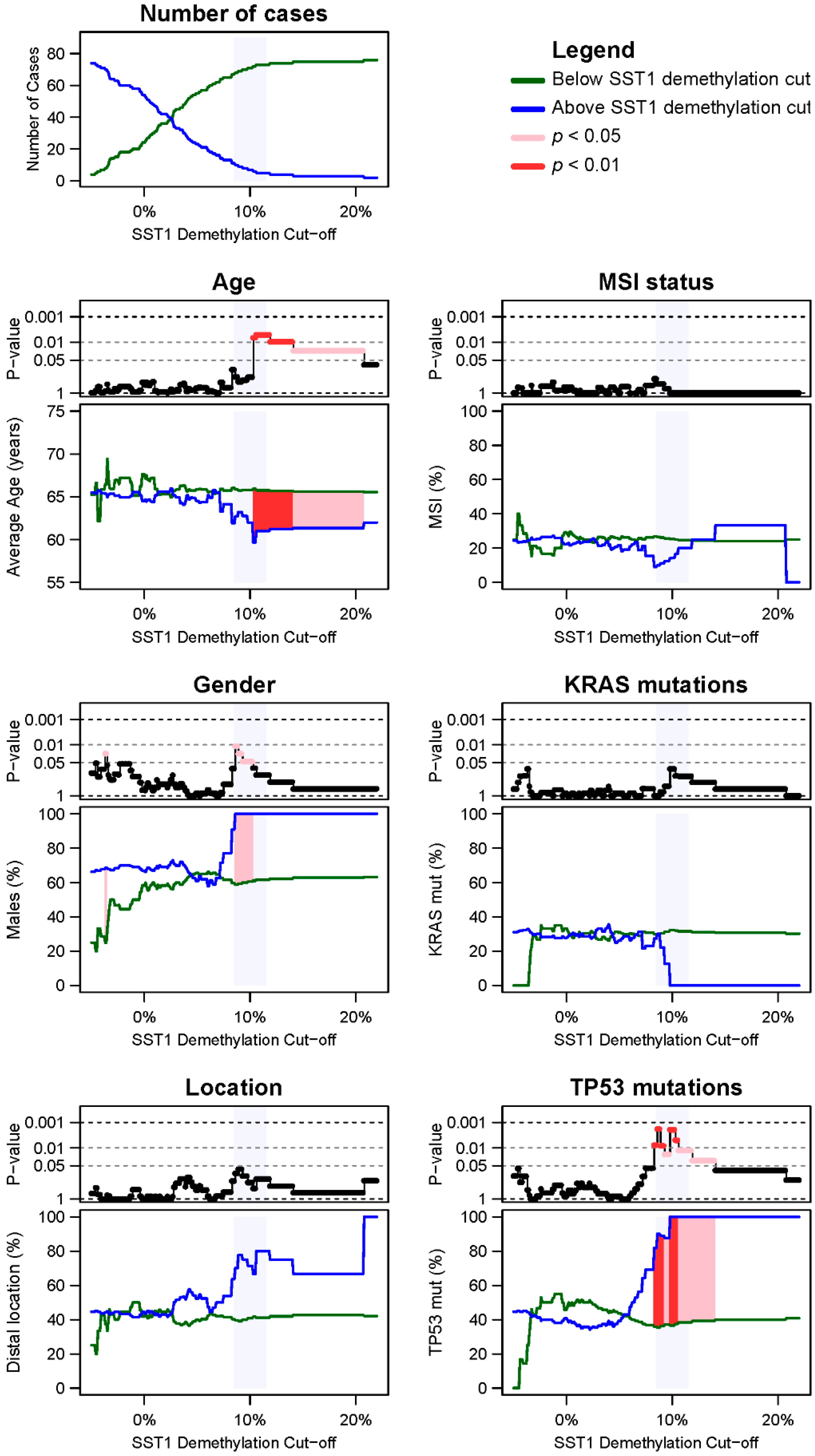
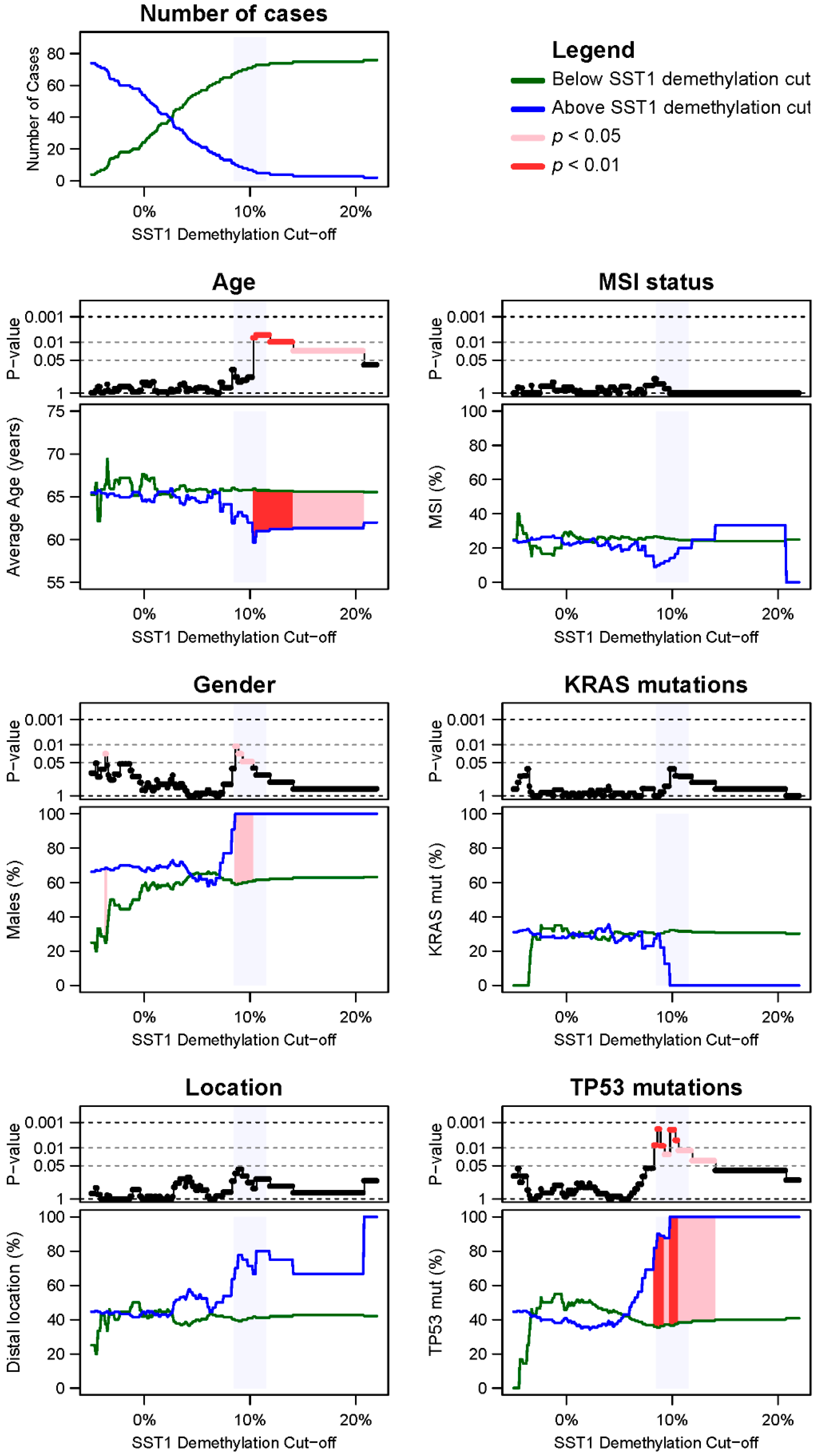
2.4. SST1 Demethylation and Genotype/Phenotype of CRC
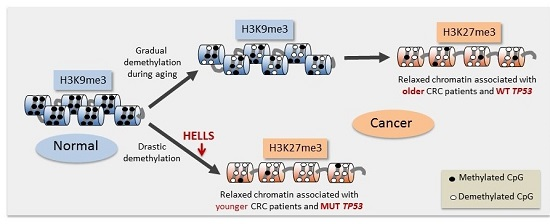
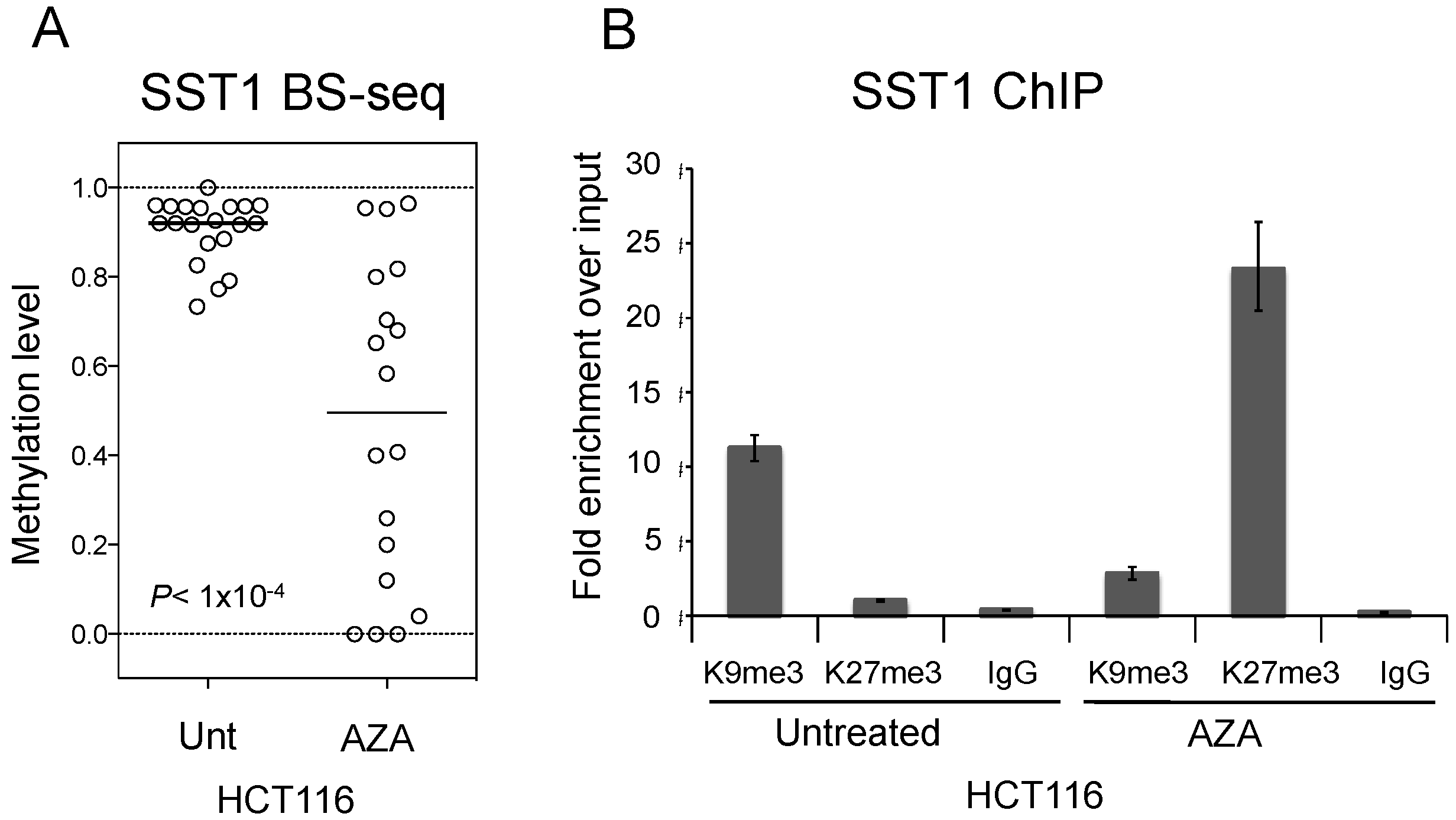
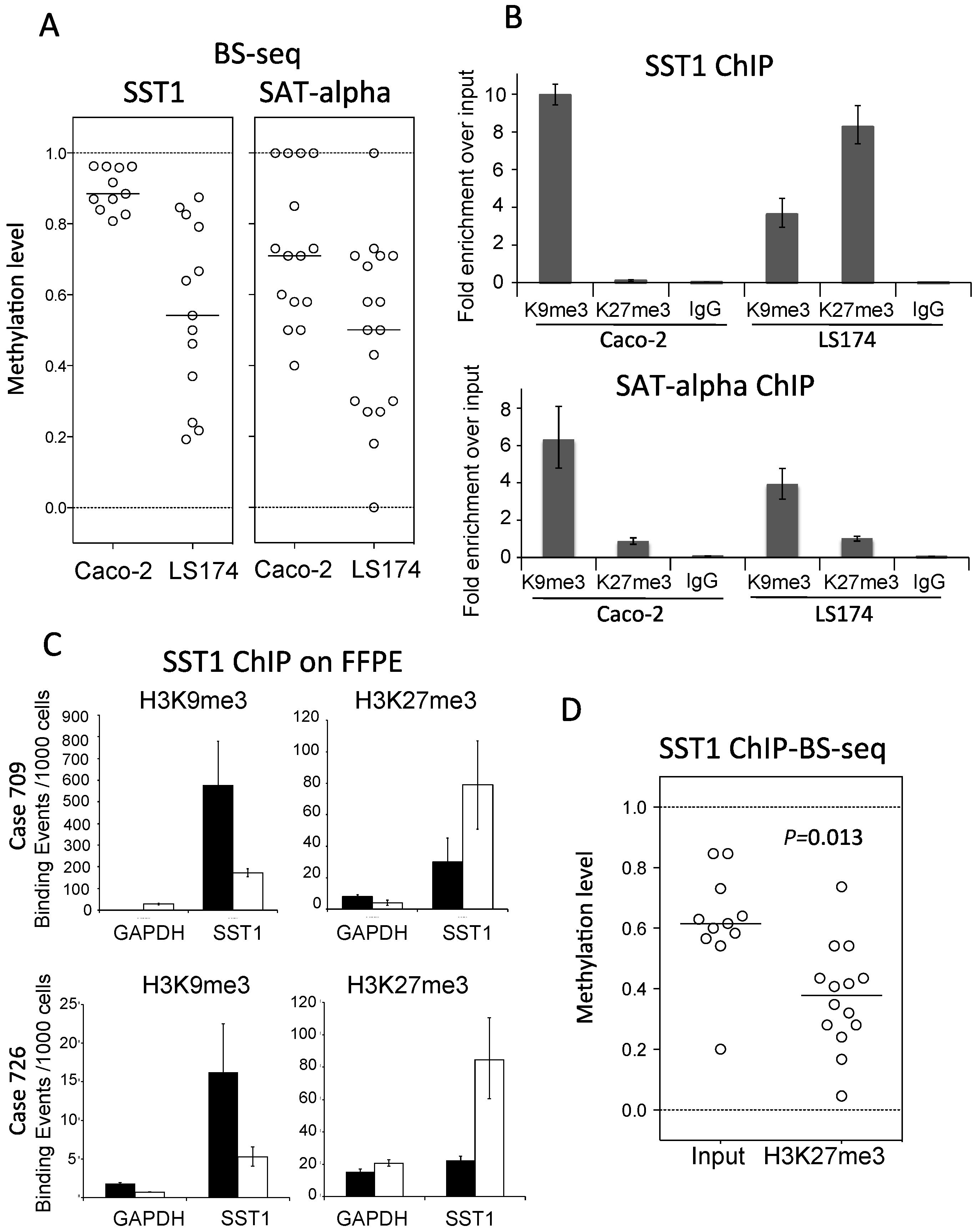
2.5. SST1 Hypomethylation Associates with Histone H3 Lysine 27 Trimethylation in Vitro and in Vivo
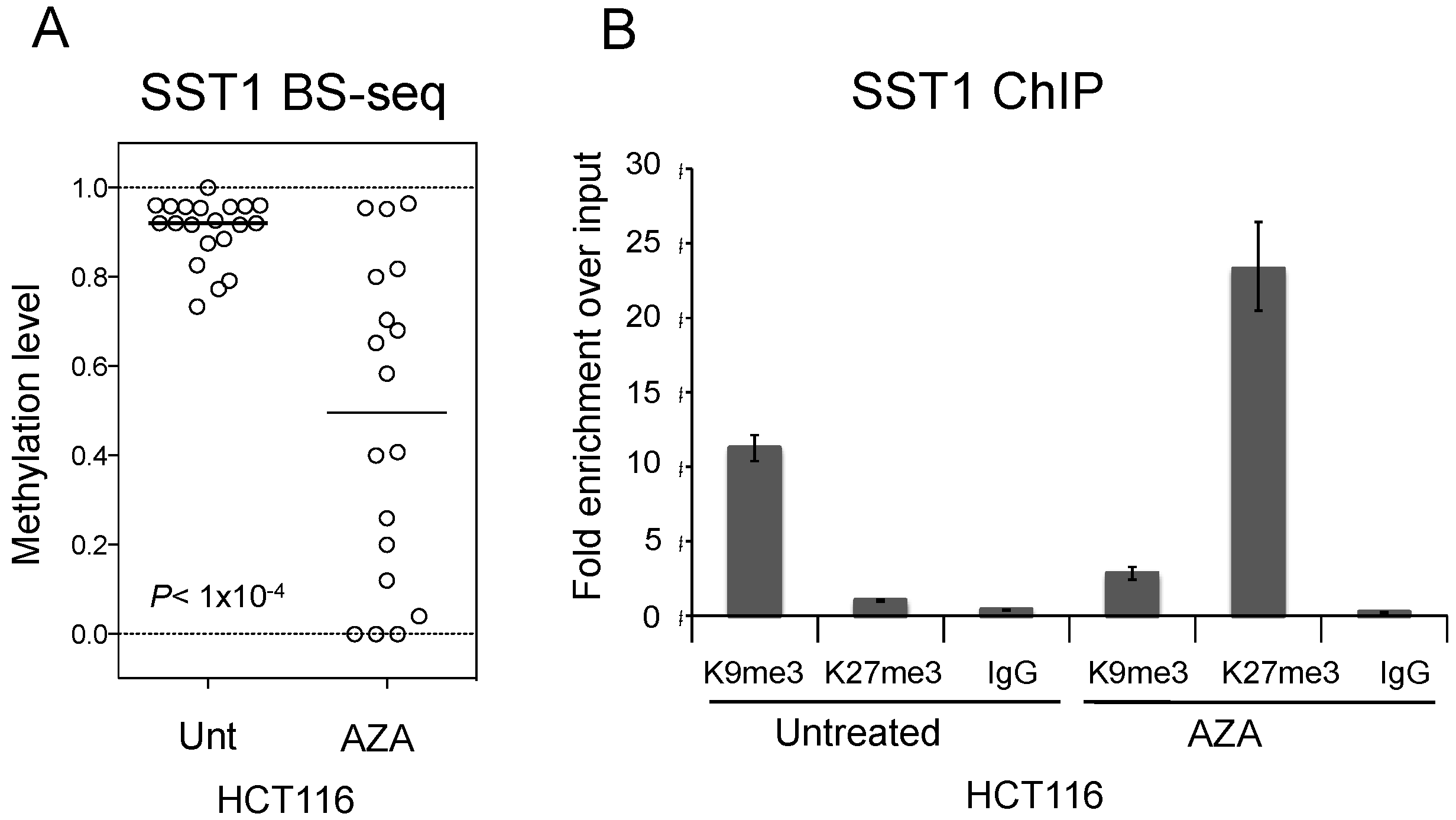
2.6. Induced SST1 Demethylation Leads to Increased H3K27me3 Levels
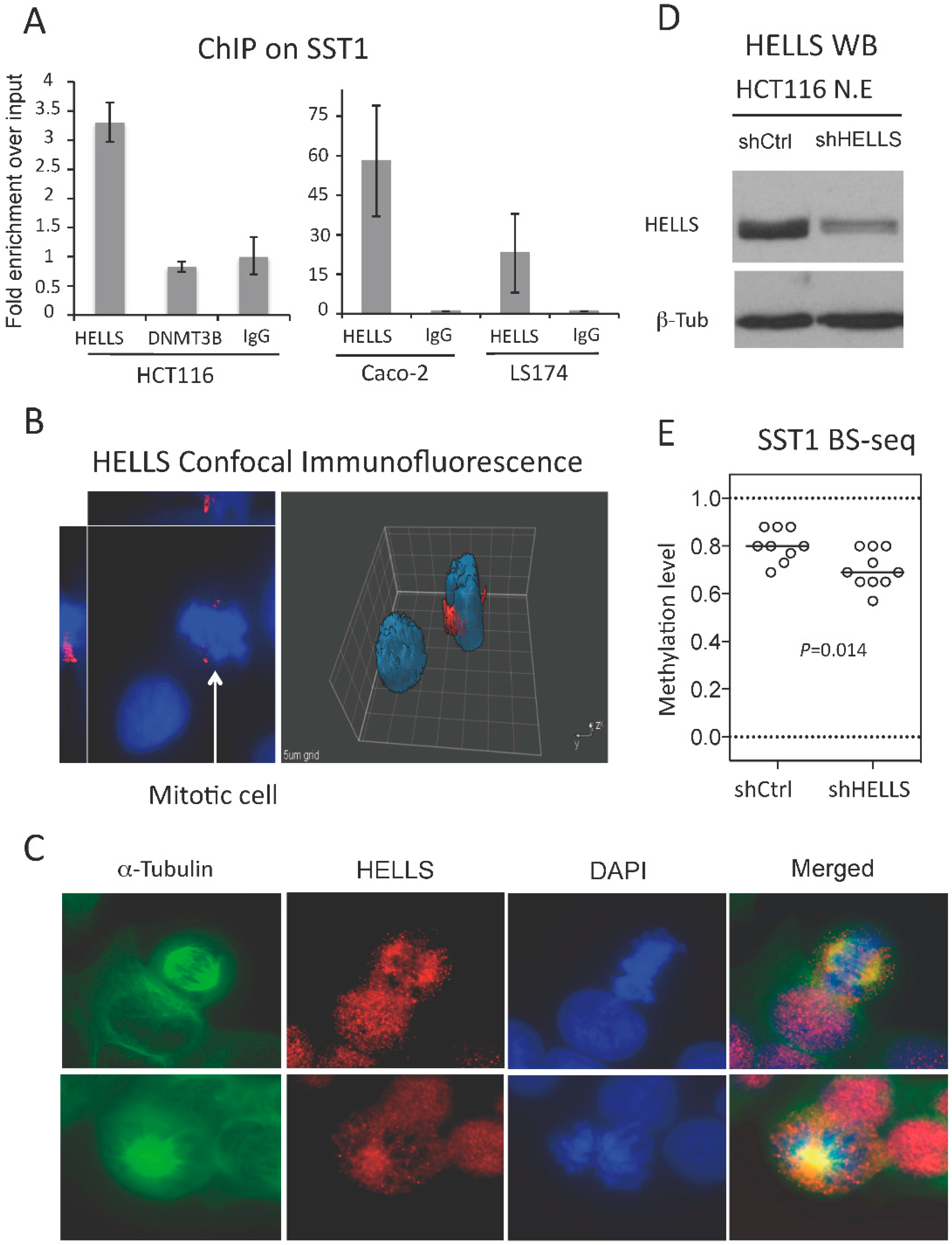
2.7. A Chromatin Remodeling Factor, HELLS, Maintains the Methylated SST1 Region
3. Discussion
4. Materials and Methods
4.1. Tissue Specimens and Samples
4.2. Genome Damage Fraction
4.3. Cancer Cell Lines and 5-aza-2′-Deoxycytidine Treatment
4.4. Molecular Analyses
4.5. Chromatin Immunoprecipitation
4.6. Immunofluorescence
4.7. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Cancer 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A new potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Schulz, W.A. Causes and consequences of DNA hypomethylation in human cancer. Biochem. Cell Biol. 2005, 83, 296–321. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.; Dubeau, L.; Narayan, A.; Yu, M.C.; Ehrlich, M. Satellite DNA hypomethylation vs. Overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutat. Res. 1999, 423, 91–101. [Google Scholar] [CrossRef]
- Wong, N.; Lam, W.C.; Lai, P.B.; Pang, E.; Lau, W.Y.; Johnson, P.J. Hypomethylation of chromosome 1 heterochromatin DNA correlates with q-arm copy gain in human hepatocellular carcinoma. Am. J. Pathol. 2001, 159, 465–471. [Google Scholar] [CrossRef]
- Widschwendter, M.; Jiang, G.; Woods, C.; Müller, H.M.; Fiegl, H.; Goebel, G.; Marth, C.; Müller-Holzner, E.; Zeimet, A.G.; Laird, P.W.; et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004, 64, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Jeanpierre, M.; Turleau, C.; Aurias, A.; Prieur, M.; Ledeist, F.; Fischer, A.; Viegas-Pequignot, E. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 1993, 2, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Kokalj-Vokac, N.; Almeida, A.; Viegas-Pequignot, E.; Jeanpierre, M.; Malfoy, B.; Dutrillaux, B. Specific induction of uncoiling and recombination by azacytidine in classical satellite-containing constitutive heterochromatin. Cytogenet. Cell Genet. 1993, 63, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Miniou, P.; Jeanpierre, M.; Blanquet, V.; Sibella, V.; Bonneau, D.; Herbelin, C.; Fischer, A.; Niveleau, A.; Viegas-Pequignot, E.T. Abnormal methylation pattern in constitutive and facultative (X inactive chromosome) heterochromatin of ICF patients. Hum. Mol. Genet. 1994, 3, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Mays-Hoopes, L.L.; Brown, A.; Huang, R.C. Methylation and rearrangement of mouse intracisternal A particle genes in development, aging, and myeloma. Mol. Cell Biol. 1983, 3, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.L.; Smith, R.A.; Ma, S.; Cutler, R.G. Genomic 5-methyldeoxycytidine decreases with age. J. Biol. Chem. 1987, 262, 9948–9951. [Google Scholar] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Jintaridth, P.; Mutirangura, A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol. Genom. 2010, 41, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, I.; Leodolter, A.; Alonso, S.; Horiuchi, S.; Yamashita, K.; Perucho, M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006, 9, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.Z.; Grundy, P.E.; Narayan, A.; Ehrlich, M. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet. Cytogenet. 1999, 109, 34–39. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: http://www.repeatmasker.org (accessed on 19 April 2012).
- Thoraval, D.; Asakawa, J.; Wimmer, K.; Kuick, R.; Lamb, B.; Richardson, B.; Ambros, P.; Glover, T.; Hanash, S. Demethylation of repetitive DNA sequences in neuroblastoma. Genes Chromosomes Cancer 1996, 17, 234–244. [Google Scholar] [CrossRef]
- Nishiyama, R.; Qi, L.; Tsumagari, K.; Weissbecker, K.; Dubeau, L.; Champagne, M.; Sikka, S.; Nagai, H.; Ehrlich, M. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol. Ther. 2005, 4, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Gehrke, C.; Kuo, K.C.; Ehrlich, M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988, 48, 1159–1161. [Google Scholar] [PubMed]
- Kazazian, H.H., Jr. L1 retrotransposons shape the mammalian genome. Science 2000, 289, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Estécio, M.R.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, 38. [Google Scholar] [CrossRef] [PubMed]
- Peinado, M.A.; Malkhosyan, S.; Velazquez, A.; Perucho, M. Isolation and characterization of allelic losses and gains in colorectal tumors by arbitrarily primed polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1992, 89, 100065–100069. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohnami, S.; Tanabe, C.; Sasaki, H.; Yasuda, J.; Katai, H.; Yoshimura, K.; Terada, M.; Perucho, M.; Yoshida, T. The genomic damage estimated by arbitrarily primed PCR DNA fingerprinting is useful for the prognosis of gastric cancer. Gastroenterology 2003, 125, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.; Fan, T.; Geiman, T.; Yan, Q.; Muegge, K. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001, 15, 2940–2944. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Geiman, T.M.; Xi, S.; Jiang, Q.; Schmidtmann, A.; Chen, T.; Li, E.; Muegge, K. Lsh is involved in de novo methylation of DNA. EMBO J. 2006, 25, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Xi, S.; Shan, J.; Maunakea, A.; Che, A.; Briones, V.; Lee, E.Y.; Geiman, T.; Huang, J.; Stephens, R.; et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc. Natl. Acad. Sci. USA 2011, 108, 5626–5631. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, R.; Baumann, C.; Fan, T.; Schmidtmann, A.; Dobrinski, I.; Muegge, K. Lsh is required for meiotic chromosome synapsis and retrotransposon silencing in female germ cells. Nat. Cell Biol. 2006, 8, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fan, T.; Yan, Q.; Zhu, H.; Fox, S.; Issaq, H.J.; Best, L.; Gangi, L.; Munroe, D.; Muegge, K. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004, 32, 5019–5028. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Kim, Y.S.; Yasuda, T.; Ohmachi, Y.; Yokouchi, H.; Monden, M.; Emi, M.; Konishi, N.; Nogami, M.; Okumura, K.; et al. A novel sperm-specific hypomethylation sequence is a demethylation hotspot in human hepatocellular carcinomas. Gene 1999, 237, 15–20. [Google Scholar] [CrossRef]
- Igarashi, S.; Suzuki, H.; Niinuma, T.; Shimizu, H.; Nojima, M.; Iwaki, H.; Nobuoka, T.; Nishida, T.; Miyazaki, Y.; Takamaru, H.; et al. A novel correlation between line-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin. Cancer Res. 2010, 16, 5114–5123. [Google Scholar] [CrossRef] [PubMed]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Cheng, A.S.; Ahmed, S.; Boumber, Y.; Charo, C.; Yamochi, T.; Urano, T.; Furukawa, K.; Kwabi-Addo, B.; et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008, 40, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M.; Park, Y.J.; Mclean, C.M.; Dokshin, G.A.; Persson, J.M.; Herman, H.; Pasini, D.; Miró, X.; Donohoe, M.E.; Lee, J.T.; et al. Antagonism between DNA and H3K27 methylation at the imprinted Rasgrf1 locus. PLoS Genet. 2008, 4, e1000145. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2005, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Hagarman, J.A.; Motley, M.P.; Kristjansdottir, K.; Soloway, P.D. Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS ONE 2013, 8, e53880. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential chip-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Muñoz, M.; Vives, L.; Frangou, C.G.; Groudine, M.; Peinado, M.A. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 19809–19814. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kanai, Y.; Ushijima, S.; Kitamura, T.; Kakizoe, T.; Hirohashi, S. DNA hypomethylation on pericentromeric satellite regions significantly correlates with loss of heterozygosity on chromosome 9 in urothelial carcinomas. J. Urol. 2005, 173, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Shao, C.; Tuck-Muller, C.M.; Sogorovic, S.; Pålsson, E.; Smeets, D.; Ehrlich, M. Interphase chromosomal abnormalities and mitotic missegregation of hypomethylated sequences in ICF syndrome cells. Chromosoma 2005, 114, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Völkel, P.; Angrand, P.-O. The control of histone lysine methylation in epigenetic regulation. Biochimie 2007, 89, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef]
- Yano, M.; Ouchida, M.; Shigematsu, H.; Tanaka, N.; Ichimura, K.; Kobayashi, K.; Inaki, Y.; Toyooka, S.; Tsukuda, K.; Shimizu, N.; et al. Tumor-specific exon creation of the HELLS/SMARCA6 gene in non-small cell lung cancer. Int. J. Cancer 2004, 112, 8–13. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network TCGA. Available online: http://cancergenome.nih.gov/ (accessed on 20 September 2016).
- Dunican, D.S.; Cruickshanks, H.A.; Suzuki, M.; Semple, C.A.; Davey, T.; Arceci, R.J.; Greally, J.; Adams, I.R.; Meehan, R.R. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function. Genome Biol. 2013, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Déjardin, J. Switching between epigenetic states at pericentromeric heterochromatin. Trends Genet. 2015, 31, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Ranade, S.; Malig, M.; Antonacci, F.; Chaisson, M.; Hon, L.; Sudmant, P.H.; Graves, T.A.; Alkan, C.; Dennis, M.Y.; et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res. 2014, 24, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013, 14, R10. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Dai, T.; Dai, Y.; Yamamoto, F.; Perucho, M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003, 4, 121–131. [Google Scholar] [CrossRef]
- Yamamoto, F.; Yamamoto, M.; Soto, J.L.; Kojima, E.; Wang, E.N.; Perucho, M.; Sekiya, T.; Yamanaka, H. Notl-Msell methylation-sensitive amplied fragment length polymorhism for DNA methylation analysis of human cancers. Electrophoresis 2001, 22, 1946–1956. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samuelsson, J.K.; Dumbovic, G.; Polo, C.; Moreta, C.; Alibés, A.; Ruiz-Larroya, T.; Giménez-Bonafé, P.; Alonso, S.; Forcales, S.-V.; Perucho, M. Helicase Lymphoid-Specific Enzyme Contributes to the Maintenance of Methylation of SST1 Pericentromeric Repeats That Are Frequently Demethylated in Colon Cancer and Associate with Genomic Damage. Epigenomes 2017, 1, 2. https://doi.org/10.3390/epigenomes1010002
Samuelsson JK, Dumbovic G, Polo C, Moreta C, Alibés A, Ruiz-Larroya T, Giménez-Bonafé P, Alonso S, Forcales S-V, Perucho M. Helicase Lymphoid-Specific Enzyme Contributes to the Maintenance of Methylation of SST1 Pericentromeric Repeats That Are Frequently Demethylated in Colon Cancer and Associate with Genomic Damage. Epigenomes. 2017; 1(1):2. https://doi.org/10.3390/epigenomes1010002
Chicago/Turabian StyleSamuelsson, Johanna K., Gabrijela Dumbovic, Cristian Polo, Cristina Moreta, Andreu Alibés, Tatiana Ruiz-Larroya, Pepita Giménez-Bonafé, Sergio Alonso, Sonia-V. Forcales, and Manuel Perucho. 2017. "Helicase Lymphoid-Specific Enzyme Contributes to the Maintenance of Methylation of SST1 Pericentromeric Repeats That Are Frequently Demethylated in Colon Cancer and Associate with Genomic Damage" Epigenomes 1, no. 1: 2. https://doi.org/10.3390/epigenomes1010002
APA StyleSamuelsson, J. K., Dumbovic, G., Polo, C., Moreta, C., Alibés, A., Ruiz-Larroya, T., Giménez-Bonafé, P., Alonso, S., Forcales, S.-V., & Perucho, M. (2017). Helicase Lymphoid-Specific Enzyme Contributes to the Maintenance of Methylation of SST1 Pericentromeric Repeats That Are Frequently Demethylated in Colon Cancer and Associate with Genomic Damage. Epigenomes, 1(1), 2. https://doi.org/10.3390/epigenomes1010002