
Population Genomics and Morphology Provide Insights into the Conservation and Diversity of Apis laboriosa

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
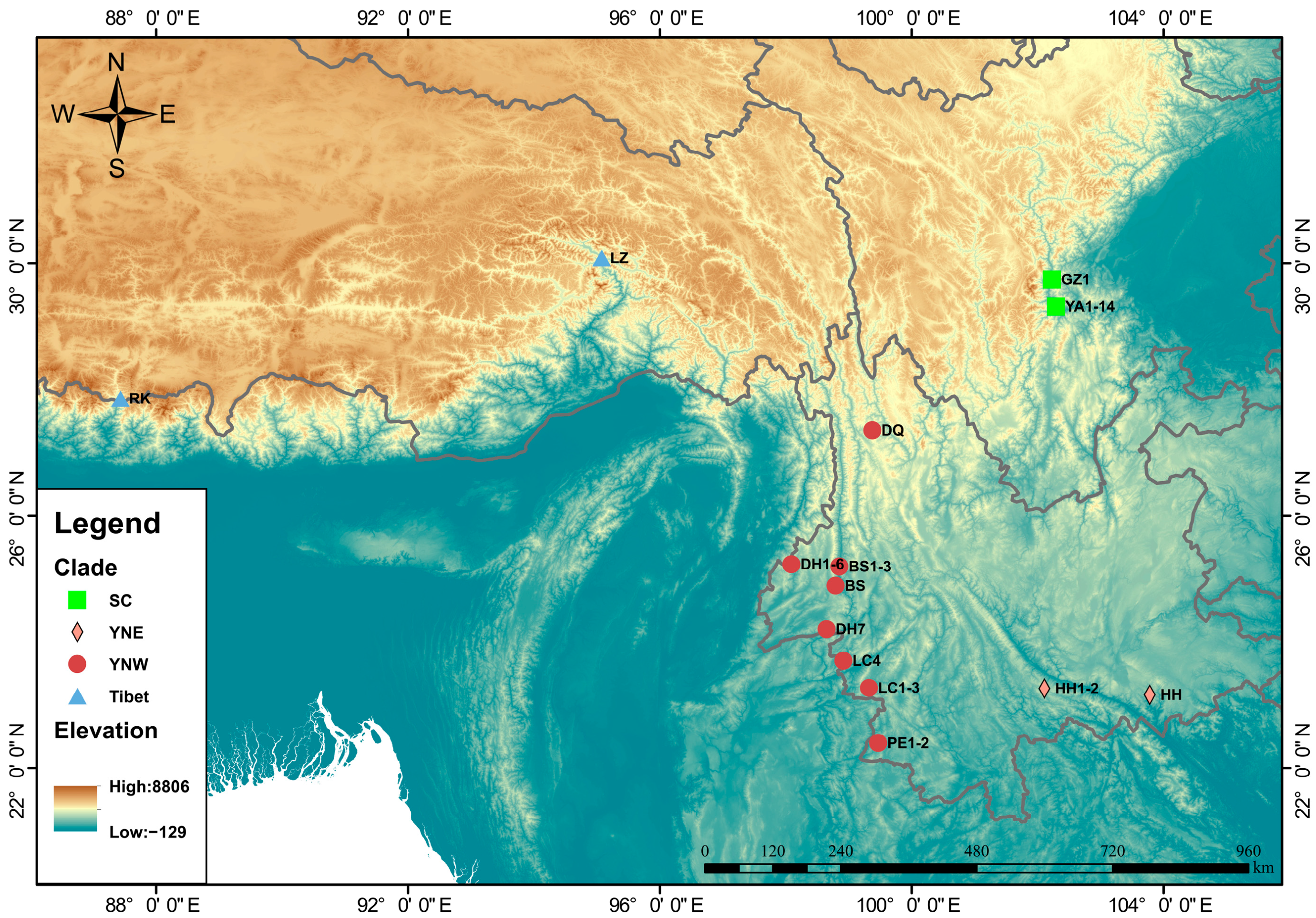
2.1. Specimen Collection and DNA Sequencing
2.2. Single-Nucleotide Polymorphism Calling
2.3. Population Structure, Principal Component Analysis, and Phylogeny Construction
2.4. Population History and Gene Flow
2.5. Mitochondrial Phylogeny
2.6. Morphological Analyses
3. Results
3.1. Population Structure
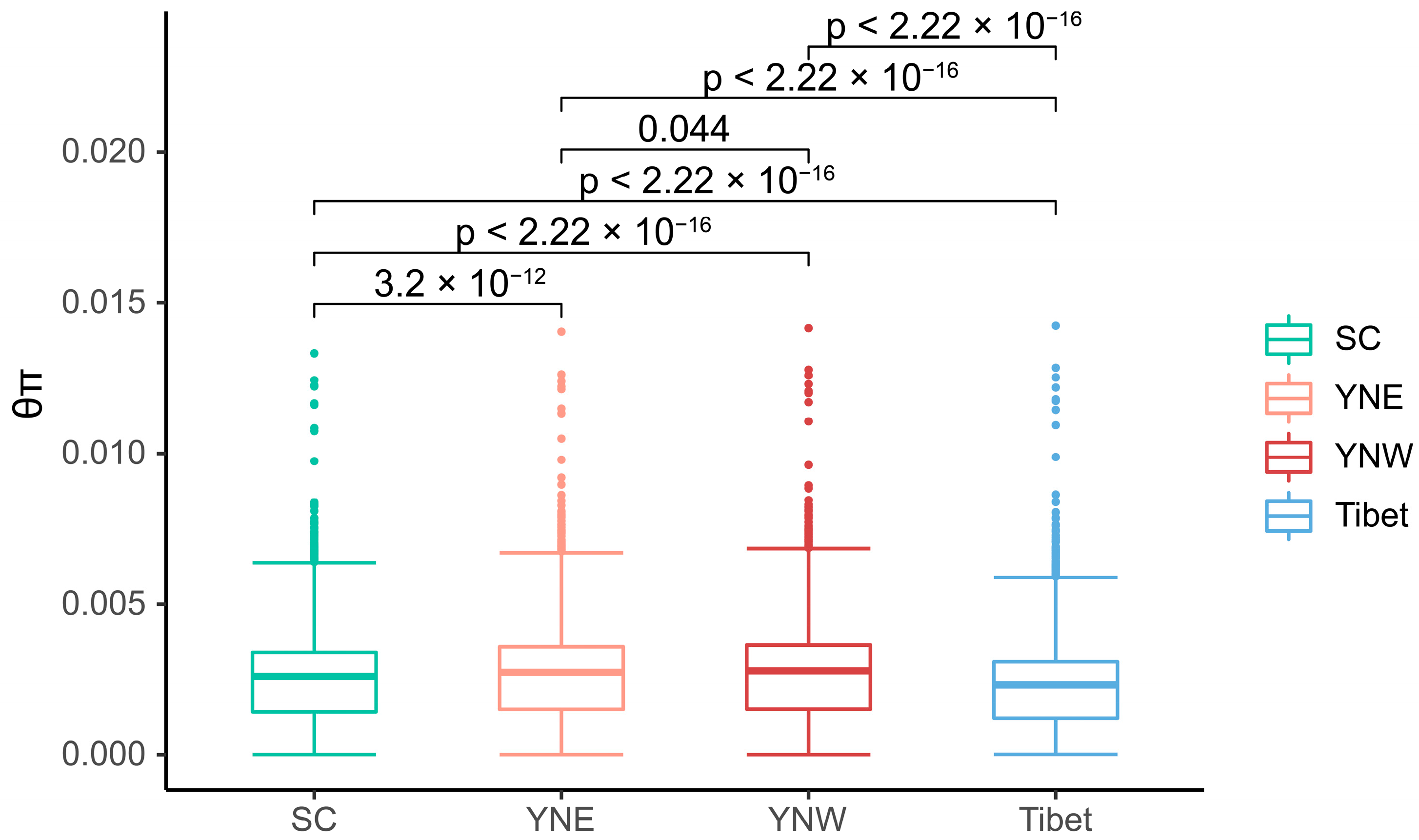
3.2. Genetic Differentiation and Genetic Diversity
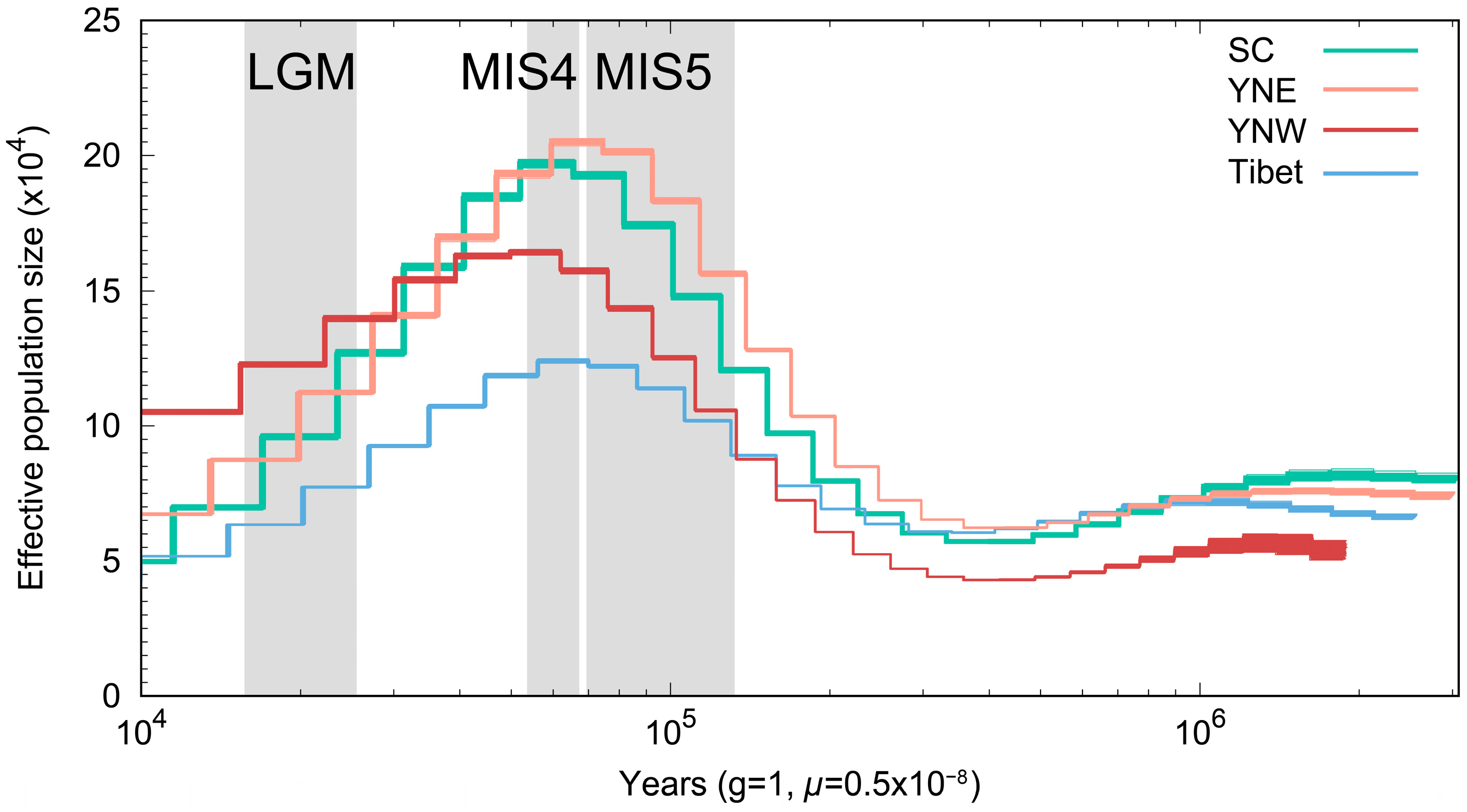
3.3. Effective Population Size Analysis
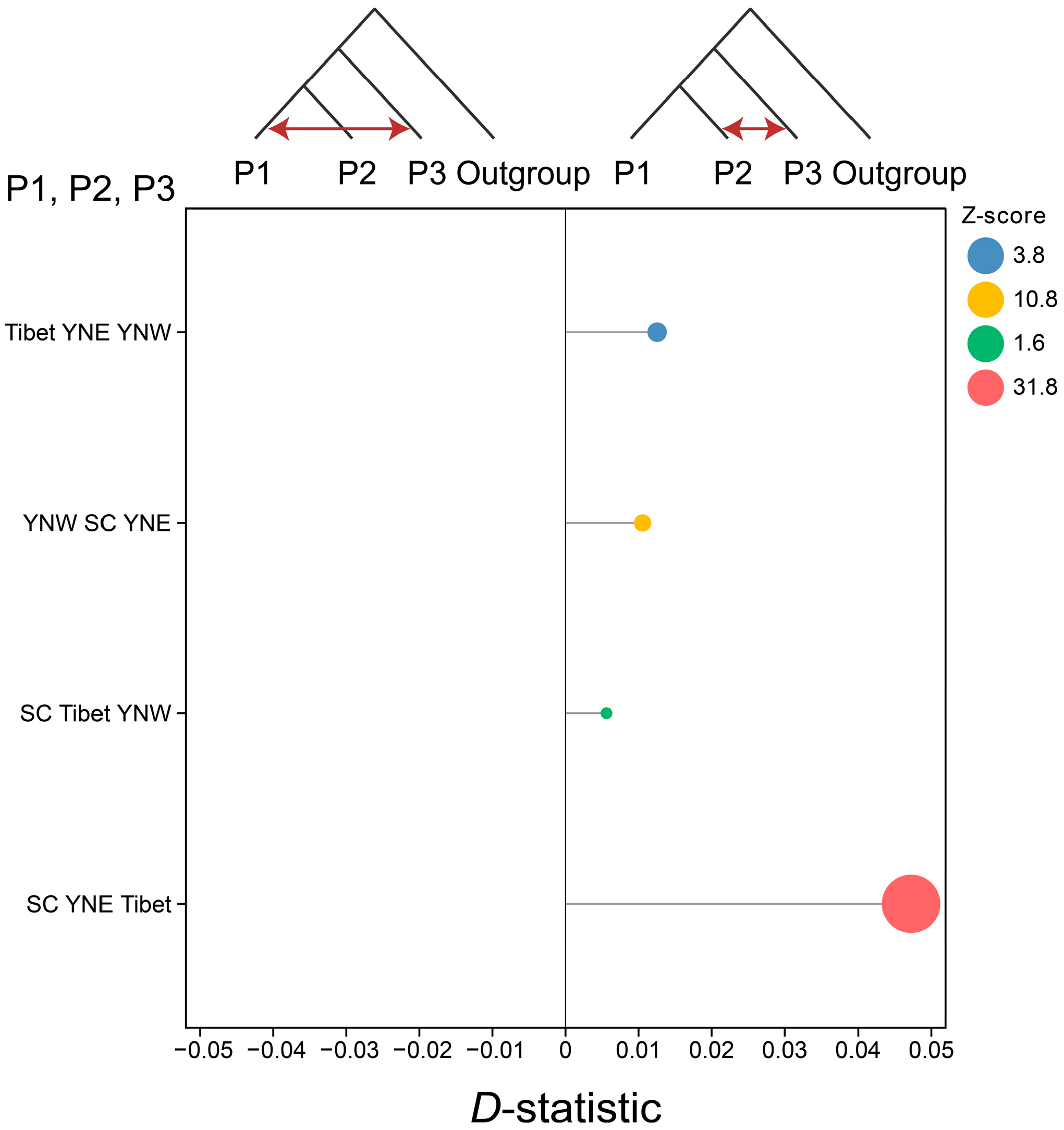
3.4. Gene Flow
3.5. Mitochondrial Phylogeny
3.6. Morphological Analyses
4. Discussion
4.1. The Status of Sichuan and Tibetan Populations
4.2. Changes in Population Size
4.3. Relationships Between Populations
4.4. Mitochondrial Phylogeny
4.5. Conservation Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rose, J.; Gilbert, M.T.P.; Outerbridge, M.; Morales, H.E. Evolutionary genomics analysis reveals a unique lineage of Megachile pruina found in an isolated population in Bermuda. Insect Conserv. Diver. 2024, 17, 1143–1155. [Google Scholar] [CrossRef]
- Qiu, L.; Dong, J.; Li, X.; Parey, S.H.; Tan, K.; Orr, M.; Majeed, A.; Zhang, X.; Luo, S.; Zhou, X.; et al. Defining honeybee subspecies in an evolutionary context warrants strategized conservation. Zool. Res. 2023, 44, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Patir, J.; Boruah, B.B.; Umbrey, C.; Tacha, K.; Taid, R.; Das, A.P.; Gogoi, H. Choice of forage plants by the giant honey bee Apis laboriosa Smith in a captive condition outside their native geographic range. J. Apicult. Res. 2023, 63, 1145–1153. [Google Scholar] [CrossRef]
- Joshi, S.R.; Ahmad, F.; Gurung, M.B. Status of Apis laboriosa populations in Kaski district, western Nepal. J. Apicult. Res. 2004, 43, 176–180. [Google Scholar] [CrossRef]
- Potts, S.G.; Imperatriz-Fonseca, V.; Ngo, H.T.; Aizen, M.A.; Biesmeijer, J.C.; Breeze, T.D.; Dicks, L.V.; Garibaldi, L.A.; Hill, R.; Settele, J.; et al. Safeguarding pollinators and their values to human well-being. Nature 2016, 540, 220–229. [Google Scholar] [CrossRef]
- Warrit, N.; Ascher, J.; Basu, P.; Belavadi, V.; Brockmann, A.; Buchori, D.; Dorey, J.B.; Hughes, A.; Krishnan, S.; Ngo, H.T.; et al. Opportunities and challenges in Asian bee research and conservation. Biol. Conserv. 2023, 285, 110173. [Google Scholar] [CrossRef]
- Engel, M.S. The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae; Apis). J. Hymenopt. Res. 1999, 8, 165–196. [Google Scholar]
- Kitnya, N.; Brockmann, A.; Otis, G.W. Taxonomic revision and identification keys for the giant honey bees. Front. Bee Sci. 2024, 2, 1379952. [Google Scholar] [CrossRef]
- Kitnya, N.; Otis, G.W.; Chakravorty, J.; Smith, D.R.; Brockmann, A. Apis laboriosa confirmed by morphometric and genetic analyses of giant honey bees (Hymenoptera, Apidae) from sites of sympatry in Arunachal Pradesh, North East India. Apidologie 2022, 53, 47. [Google Scholar] [CrossRef]
- Kuang, B.; Li, Y.; Liu, Q.; Wang, H. Morphometric analysis of two species of large honey bees (worker bees) in China. Apicult. China 1983, 4, 1–2. [Google Scholar]
- Tang, X.Y.; Yao, Y.X.; Li, Y.H.; Song, H.L.; Luo, R.; Shi, P.; Zhou, Z.Y.; Xu, J.S. Comparison of the mitochondrial genomes of three geographical strains of Apis laboriosa indicates high genetic diversity in the black giant honeybee (Hymenoptera: Apidae). Ecol. Evol. 2023, 13, e9782. [Google Scholar] [CrossRef]
- Cao, L.; Dai, Z.; Tan, H.; Zheng, H.; Wang, Y.; Chen, J.; Kuang, H.; Chong, R.A.; Han, M.; Hu, F.; et al. Population Structure, Demographic History, and Adaptation of Giant Honeybees in China Revealed by Population Genomic Data. Genome Biol. Evol. 2023, 15, evad025. [Google Scholar] [CrossRef]
- Huang, M.J.; Hughes, A.C.; Xu, C.Y.; Miao, B.; Gao, J.; Peng, Y. Mapping the changing distribution of two important pollinating giant honeybees across 21,000 years. Glob. Ecol. Conserv. 2022, 39, e02282. [Google Scholar]
- Kitnya, N.; Prabhudev, M.V.; Bhatta, C.P.; Pham, T.H.; Nidup, T.; Megu, K.; Chakravorty, J.; Brockmann, A.; Otis, G.W. Geographical distribution of the giant honey bee Apis laboriosa Smith, 1871 (Hymenoptera, Apidae). Zookeys 2020, 951, 67–81. [Google Scholar] [CrossRef]
- Voraphab, I.; Chatthanabun, N.; Nalinrachatakan, P.; Thanoosing, C.; Traiyasut, P.; Kunsete, C.; Deowanish, S.; Otis, G.W.; Warrit, N. Discovery of the Himalayan giant honey bee, Apis laboriosa, in Thailand: A major range extension. Apidologie 2024, 55, 31. [Google Scholar] [CrossRef]
- Otis, G.W.; Huang, M.; Kitnya, N.; Sheikh, U.A.A.; Faiz, A.U.H.; Phung, C.H.; Warrit, N.; Peng, Y.; Zhou, X.; Oo, H.M.; et al. The distribution of Apis laboriosa revisited: Range extensions, biogeographic affinities, and species distribution modelling. Front. Bee Sci. 2024, 2, 1374852. [Google Scholar] [CrossRef]
- Ji, Y.; Li, X.; Ji, T.; Tang, J.; Qiu, L.; Hu, J.; Dong, J.; Luo, S.; Liu, S.; Frandsen, P.B.; et al. Gene reuse facilitates rapid radiation and independent adaptation to diverse habitats in the Asian honeybee. Sci. Adv. 2020, 6, eabd3590. [Google Scholar] [CrossRef]
- Lin, D.; Lan, L.; Zheng, T.; Shi, P.; Xu, J.; Li, J. Comparative genomics reveals recent adaptive evolution in Himalayan giant honeybee Apis laboriosa. Genome Biol. Evol. 2021, 13, evab227. [Google Scholar] [CrossRef]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Russell, P.; Meyer, M.; Mauceli, E.; Alföldi, J.; Di Palma, F.; Lindblad-Toh, K. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 2010, 26, 1145–1151. [Google Scholar] [CrossRef]
- Yun, S.; Yun, S. Masking as an effective quality control method for next-generation sequencing data analysis. BMC Bioinform. 2014, 15, 382. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations, Volume 4: Variability Within and Among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Terhorst, J.; Kamm, J.A.; Song, Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017, 49, 303–309. [Google Scholar] [CrossRef]
- Wallberg, A.; Han, F.; Wellhagen, G.; Dahle, B.; Kawata, M.; Haddad, N.; Simoes, Z.L.; Allsopp, M.H.; Kandemir, I.; De la Rua, P.; et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 2014, 46, 1081–1088. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Takahashi, J.; Rai, J.; Wakamiya, T.; Okuyama, H. Characterization of the complete mitochondrial genome of the giant black Himalayan honeybee (Apis laboriosa) from Nepal. Conserv. Genet. Resour. 2018, 10, 59–63. [Google Scholar] [CrossRef]
- Wang, A.R.; Kim, J.S.; Kim, M.J.; Kim, H.; Choi, Y.S.; Kim, I. Comparative description of mitochondrial genomes of the honey bee Apis (Hymenoptera: Apidae): Four new genome sequences and Apis phylogeny using whole genomes and individual genes. J. Apicult. Res. 2018, 57, 484–503. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin, Germany, 1988. [Google Scholar]
- Dogantzis, K.A.; Tiwari, T.; Conflitti, I.M.; Dey, A.; Patch, H.M.; Muli, E.M.; Garnery, L.; Whitfield, C.W.; Stolle, E.; Alqarni, A.S.; et al. Thrice out of Asia and the adaptive radiation of the western honey bee. Sci. Adv. 2021, 7, eabj2151. [Google Scholar] [CrossRef]
- Helmens, K.F. The Last Interglacial–Glacial cycle (MIS 5–2) re-examined based on long proxy records from central and northern Europe. Quat. Sci. Rev. 2014, 86, 115–143. [Google Scholar] [CrossRef]
- Jouzel, J.; Masson-Delmotte, V.; Cattani, O.; Dreyfus, G.; Falourd, S.; Hoffmann, G.; Minster, B.; Nouet, J.; Barnola, J.M.; Chappellaz, J.; et al. Orbital and Millennial Antarctic Climate Variability over the Past 800,000 Years. Science 2007, 317, 793–796. [Google Scholar] [CrossRef]
- Lisiecki, L.E.; Raymo, M.E. A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 2005, 20, PA1003. [Google Scholar]
- Chen, C.; Wang, H.; Liu, Z.; Chen, X.; Tang, J.; Meng, F.; Shi, W. Population genomics provide insights into the evolution and adaptation of the eastern honey bee (Apis cerana). Mol. Biol. Evol. 2018, 35, 2260–2271. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The Last Glacial Maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef]
- Hagan, T.; Ding, G.; Buchmann, G.; Oldroyd, B.P.; Gloag, R. Serial founder effects slow range expansion in an invasive social insect. Nat. Commun. 2024, 15, 3608. [Google Scholar] [CrossRef]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef]
- Sun, S.Q.; Wu, Y.H.; Wang, G.X.; Zhou, J.; Yu, D.; Bing, H.J.; Luo, J. Bryophyte species richness and composition along an altitudinal gradient in Gongga Mountain, China. PLoS ONE 2013, 8, e58131. [Google Scholar] [CrossRef]
- Barr, C.M.; Neiman, M.; Taylor, D.R. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 2005, 168, 39–50. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Liu, S.; Donath, A.; Peters, R.S.; Ware, J.; Misof, B.; Niehuis, O.; Pfrender, M.E.; Zhou, X. The molecular evolutionary dynamics of oxidative phosphorylation (OXPHOS) genes in Hymenoptera. BMC Evol. Biol. 2017, 17, 269. [Google Scholar] [CrossRef]
- Kazenel, M.R.; Wright, K.W.; Griswold, T.; Whitney, K.D.; Rudgers, J.A. Heat and desiccation tolerances predict bee abundance under climate change. Nature 2024, 628, 342–348. [Google Scholar] [CrossRef]
- Basavarajappa, S. Studies on the impact of anthropogenic interference on wild honeybees in Mysore District, Karnataka, India. Afr. J. Agric. Res. 2010, 5, 298–305. [Google Scholar]
- Herrera, C.M. Gradual replacement of wild bees by honeybees in flowers of the Mediterranean Basin over the last 50 years. Proc. R. Soc. B 2020, 287, 20192657. [Google Scholar] [CrossRef]
- Tourbez, C.; Fiordaliso, W.; Bar-Massada, A.; Dolev, A.; Michez, D.; Dorchin, A. Commercial honey bee keeping compromises wild bee conservation in Mediterranean nature reserves. Apidologie 2025, 56, 11. [Google Scholar] [CrossRef]
- Pasquali, L.; Bruschini, C.; Benetello, F.; Bonifacino, M.; Giannini, F.; Monterastelli, E.; Penco, M.; Pesarini, S.; Salvati, V.; Simbula, G.; et al. Island-wide removal of honeybees reveals exploitative trophic competition with strongly declining wild bee populations. Curr. Biol. 2025, 35, 1576–1590.e12. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Ma, X.; Zhang, L.; Lai, K.; Shu, C.; Wang, B.; Zhang, M.; Yang, M. Population Genomics and Morphology Provide Insights into the Conservation and Diversity of Apis laboriosa. Insects 2025, 16, 546. https://doi.org/10.3390/insects16050546
Liu R, Ma X, Zhang L, Lai K, Shu C, Wang B, Zhang M, Yang M. Population Genomics and Morphology Provide Insights into the Conservation and Diversity of Apis laboriosa. Insects. 2025; 16(5):546. https://doi.org/10.3390/insects16050546
Chicago/Turabian StyleLiu, Ri, Xuntao Ma, Longfu Zhang, Kang Lai, Changbin Shu, Bin Wang, Mingwang Zhang, and Mingxian Yang. 2025. "Population Genomics and Morphology Provide Insights into the Conservation and Diversity of Apis laboriosa" Insects 16, no. 5: 546. https://doi.org/10.3390/insects16050546
APA StyleLiu, R., Ma, X., Zhang, L., Lai, K., Shu, C., Wang, B., Zhang, M., & Yang, M. (2025). Population Genomics and Morphology Provide Insights into the Conservation and Diversity of Apis laboriosa. Insects, 16(5), 546. https://doi.org/10.3390/insects16050546