
Morpho-Molecular Discordance and Cryptic Diversity in Jumping Bristletails: A Mitogenomic Analysis of Pedetontus silvestrii (Insecta: Archaeognatha: Machilidae)


 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Species Collection, Morphological Identification, and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Mitogenome Annotation and Sequencing Analysis
2.4. Phylogenetic Analyses
2.5. Divergence Time Estimation
3. Results
3.1. Composition of Mitogenomes
3.2. Estimation and Evaluation of Genetic Distance
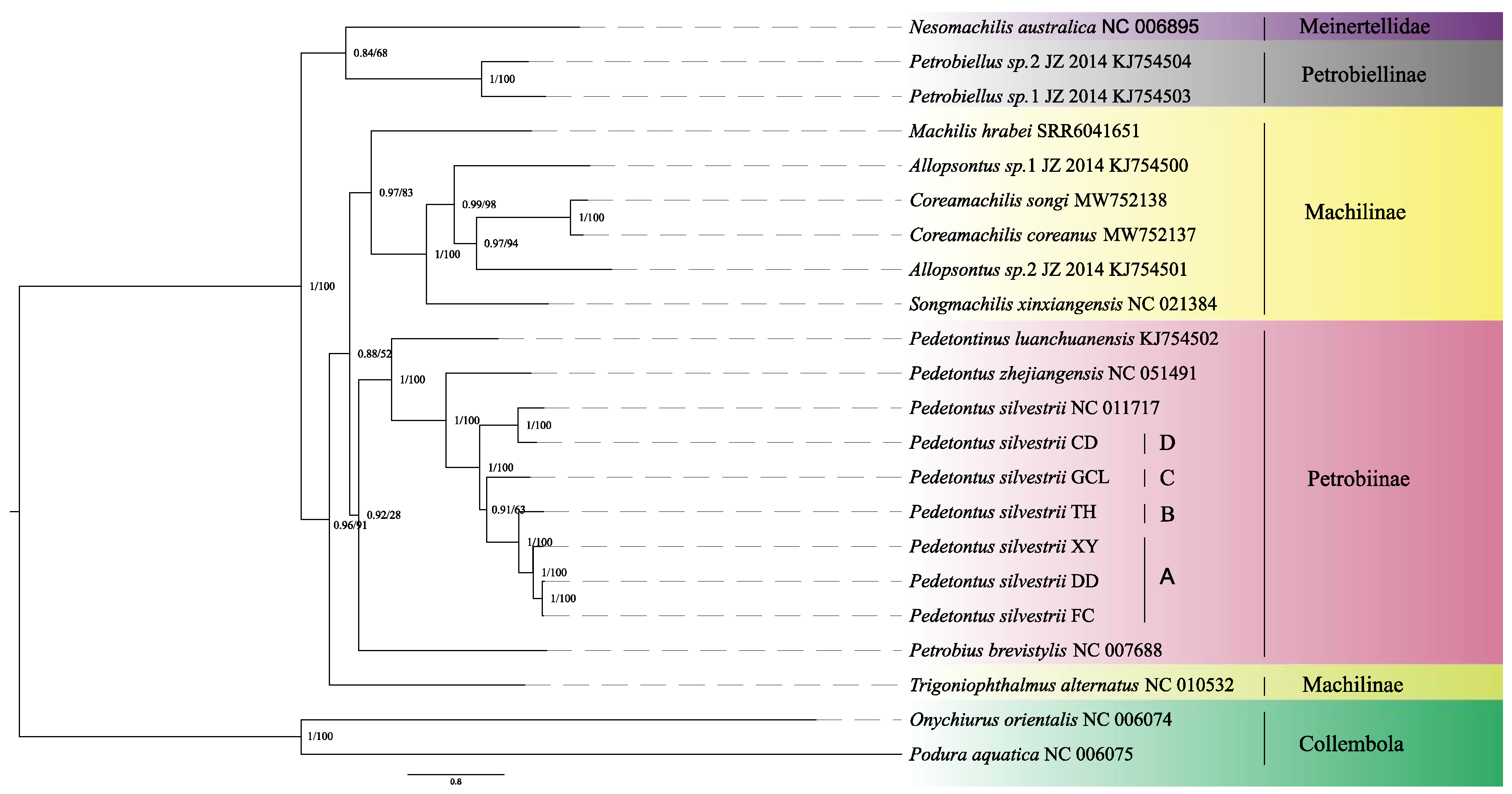
3.3. Phylogenetic Relationship
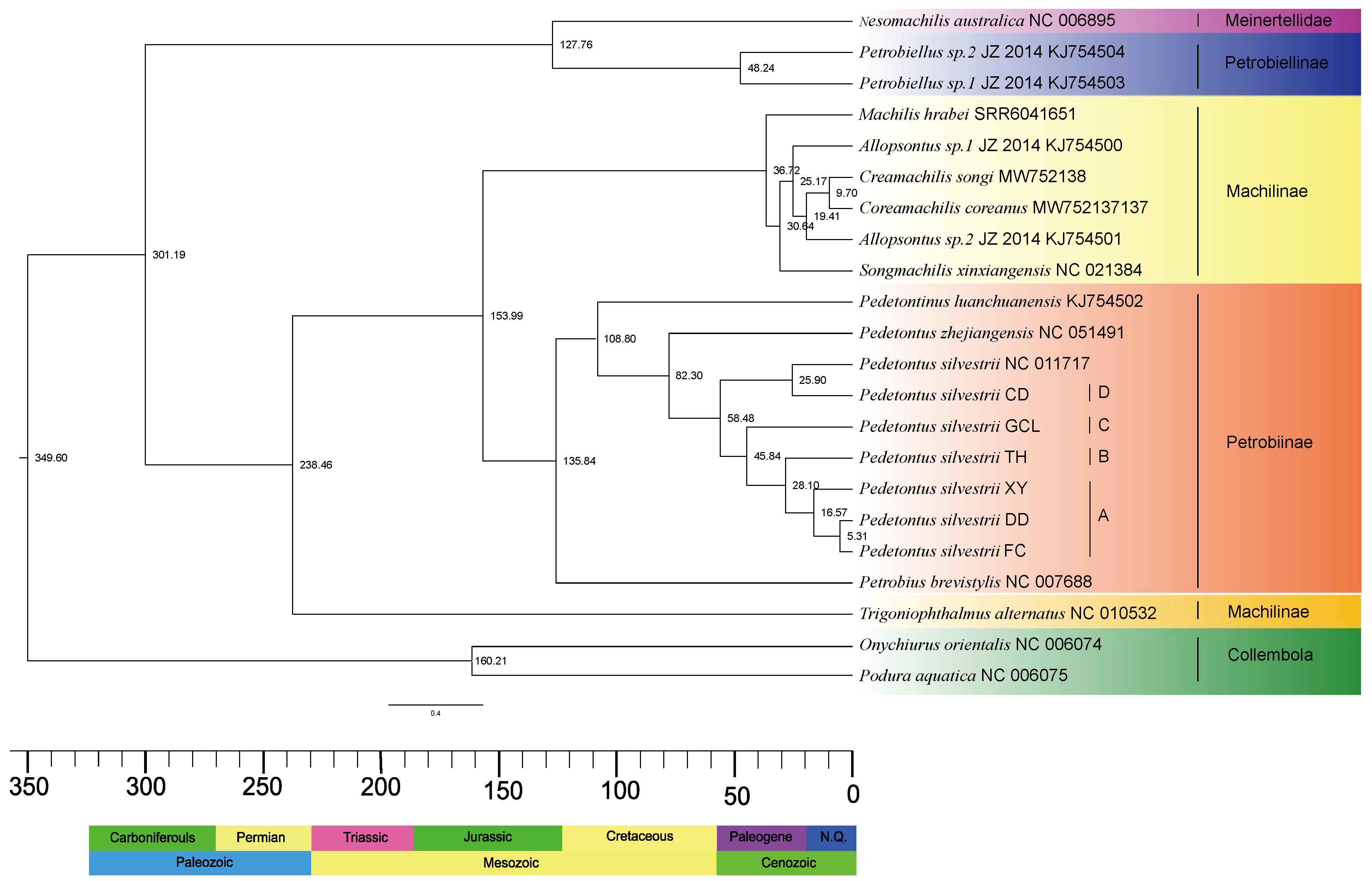
3.4. Estimation of Divergence Time
4. Discussion
4.1. Comparison of Mitogenome Composition
4.2. Phylogenetic Relationship and Divergence Time of Archaeognatha
4.3. Identification of Cryptic Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sturm, H.; Bach, D.R.C. On the systematics of the Archaeognatha (Insecta). Entomol. Gen. 1993, 18, 55–90. [Google Scholar] [CrossRef]
- Zhang, W.; Li, H.; Shih, C.; Zhang, A.; Ren, D. Phylogenetic analyses with four new Cretaceous bristletails reveal inter-relationships of Archaeognatha and Gondwana origin of Meinertellidae. Cladistics 2018, 34, 384–406. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, D.A. 400 million years on six legs: On the origin and early evolution of Hexapoda. Arthropod Struct. Dev. 2010, 39, 191–203. [Google Scholar] [CrossRef]
- Tremmel, M.; Müller, C. Insect personality depends on environmental conditions. Behav. Ecol. 2013, 24, 386–392. [Google Scholar] [CrossRef]
- Ma, Y.; He, K.; Yu, P.P.; Yu, D.N.; Cheng, X.F.; Zhang, J.Y. The complete mitochondrial genomes of three bristletails (Insecta: Archaeognatha): The paraphyly of Machilidae and insights into Archaeognathan phylogeny. PLoS ONE 2015, 10, e0117669. [Google Scholar] [CrossRef]
- He, K.; Zhang, J.Y.; Deng, K.Z.; Chen, Z. The complete mitochondrial genome of the bristletail Songmachilis xinxiangensis (Archaeognatha: Machilidae). Mitochondr. DNA 2013, 24, 99–101. [Google Scholar] [CrossRef]
- Łukasik, P.; Chong, R.A.; Nazario, K.; Matsuura, Y.; Bublitz, D.A.C.; Campbell, M.A.; Meyer, M.C.; Van Leuven, J.T.; Pessacq, P.; Veloso, C. One hundred mitochondrial genomes of cicadas. J. Hered. 2019, 110, 247–256. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef]
- Guo, Z.Q.; Shen, C.Y.; Cheng, H.Y.; Chen, Y.X.; Wu, H.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Mitogenome-based phylogeny with divergence time estimates revealed the presence of cryptic species within Heptageniidae (Insecta, Ephemeroptera). Insects 2024, 15, 745. [Google Scholar] [CrossRef]
- Boore, J.L. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol. Evol. 2006, 21, 439–446. [Google Scholar] [CrossRef]
- Tong, Y.; Shen, C.Y.; Zhao, Y.Y.; Lin, Y.J.; Wu, L.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The genetic diversity and the divergence time in extant primitive mayfly, Siphluriscus chinensis Ulmer, 1920 using the mitochondrial genome. Genes 2022, 13, 1780. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, H.; Zhao, T. Insights into the phylogeny of Hemiptera from increased mitogenomic taxon sampling. Mol. Phylogenet. Evol. 2019, 137, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Sweet, A.D.; Johnson, K.P.; Cao, Y.; de Moya, R.S.; Skinner, R.K.; Tan, M.; Herrera, S.V.; Cameron, S.L. Structure, gene order, and nucleotide composition of mitochondrial genomes in parasitic lice from Amblycera. Gene 2021, 768, 145312. [Google Scholar] [CrossRef]
- Hotaling, S.; Sproul, J.S.; Heckenhauer, J.; Powell, A.; Larracuente, A.M.; Pauls, S.U.; Kelley, J.L.; Frandsen, P.B. Long reads are revolutionizing 20 years of insect genome sequencing. Genome Biol. Evol. 2021, 13, evab138. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: A decade of progress. Annu. Rev. Entomol. 2024, 70, 83–101. [Google Scholar] [CrossRef]
- Linard, B.; Crampton-Platt, A.; Gillett, C.P.; Timmermans, M.J.; Vogler, A.P. Metagenome skimming of insect specimen pools: Potential for comparative genomics. Genome Biol. Evol. 2015, 7, 1474–1489. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhou, K.Y. Descriptions of one new genus and six new species of Machilidae (Insecta: Archaeognatha) from China: Morphological and molecular data. J. Nat. Hist. 2011, 45, 1131–1164. [Google Scholar] [CrossRef]
- Mendes, L. New contribution towards the knowledge of the Northern Korean thysanurans (Microcoryphia and Zygentoma: Insecta). Garcia Orta 1991, 18, 67–78. [Google Scholar]
- Notario-Muñoz, M.J.; de Roca, C.B.; Gaju-Ricart, M. Machilinus costai, a new species of Meinertellidae (Insecta, Microcoryphia) from Spain. Pedobiologia 2000, 44, 300–308. [Google Scholar] [CrossRef]
- Remington, C.L. The suprageneric classification of the order Thysanura (Insecta). Ann. Entomol. Soc. Am. 1954, 47, 277–286. [Google Scholar] [CrossRef]
- Gaju-Ricart, M.; Baltanás, R.M.; de Roca, C.B. Forward without wings: Current progress and future perspectives in the study of Microcoryphia and Zygentoma. Soil Org. 2015, 87, 183–195. [Google Scholar]
- Giribet, G.; Edgecombe, G.D.; Carpenter, J.M.; D’Haese, C.A.; Wheeler, W.C. Is Ellipura monophyletic? A combined analysis of basal hexapod relationships with emphasis on the origin of insects. Org. Divers. Evol. 2004, 4, 319–340. [Google Scholar] [CrossRef]
- Dell’Ampio, E.; Szucsich, N.U.; Carapelli, A.; Frati, F.; Steiner, G.; Steinacher, A.; Pass, G. Testing for misleading effects in the phylogenetic reconstruction of ancient lineages of hexapods: Influence of character dependence and character choice in analyses of 28S rRNA sequences. Zool. Scr. 2009, 38, 155–170. [Google Scholar] [CrossRef]
- Bechly, G.; Stockar, R. The first Mesozoic record of the extinct apterygote insect genus Dasyleptus (Insecta: Archaeognatha: Monura: Dasyleptidae) from the Triassic of Monte San Giorgio (Switzerland). Palaeodiversity 2011, 4, 23–37. [Google Scholar]
- Korshunova, T.; Picton, B.; Furfaro, G.; Mariottini, P.; Pontes, M.; Prkić, J.; Fletcher, K.; Malmberg, K.; Lundin, K.; Martynov, A. Multilevel fine-scale diversity challenges the ‘cryptic species’ concept. Sci. Rep. 2019, 9, 6732. [Google Scholar] [CrossRef]
- Tong, Y.; Wu, L.; Ayivi, S.P.G.; Storey, K.B.; Ma, Y.; Yu, D.N.; Zhang, J.Y. Cryptic species exist in Vietnamella sinensis Hsu, 1936 (Insecta: Ephemeroptera) from studies of complete mitochondrial genomes. Insects 2022, 13, 412. [Google Scholar] [CrossRef]
- Yang, Y.M.; Zhang, L.H.; Lin, Y.J.; Zheng, Y.M.; Jin, W.T.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The genetic diversity in Thereuonema tuberculata (Wood, 1862) (Scutigeromorpha: Scutigeridae) and the phylogenetic relationship of Scutigeromorpha using the mitochondrial genome. Insects 2022, 13, 620. [Google Scholar] [CrossRef]
- Ge, J.J.; Ying, H.F.; Xu, S.Q.; Huang, H.T. Mitochondrial genome phylogeny reveals the deep-time origin of Gomphomastacinae (Orthoptera: Eumastacidae) and its alpine genera in China. J. Syst. Evol. 2023, 61, 1047–1055. [Google Scholar] [CrossRef]
- Pakrashi, A.; Kumar, V.; Stanford-Beale, D.A.; Cameron, S.L.; Tyagi, K. Gene arrangement, phylogeny and divergence time estimation of mitogenomes in Thrips. Mol. Biol. Rep. 2022, 49, 6269–6283. [Google Scholar] [CrossRef]
- Wang, L.; Ding, S.; Cameron, S.L.; Li, X.; Liu, Y.; Yao, G.; Yang, D. Middle Jurassic origin in India: A new look at evolution of Vermileonidae and time-scaled relationships of lower brachyceran flies. Zool. J. Linn. Soc. 2022, 194, 938–959. [Google Scholar] [CrossRef]
- Shear, W.A.; Bonamo, P.M.; Grierson, J.D.; Rolfe, W.I.; Smith, E.L.; Norton, R.A. Early land animals in North America: Evidence from Devonian age arthropods from Gilboa, New York. Science 1984, 224, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Labandeira, C.C.; Beall, B.S.; Hueber, F.M. Early insect diversification: Evidence from a Lower Devonian bristletail from Québec. Science 1988, 242, 913–916. [Google Scholar] [CrossRef]
- Grimaldi, D. Insect evolutionary history from Handlirsch to Hennig, and beyond. J. Paleontol. 2001, 75, 1152–1160. [Google Scholar] [CrossRef]
- Getty, P.R.; Sproule, R.; Wagner, D.L.; Bush, A.M. Variation in wingless insect trace fossils: Insights from neoichnology and the Pennsylvanian of Massachussetts. Palaios 2013, 28, 243–258. [Google Scholar] [CrossRef]
- Sturm, H.; Poinar, G.O., Jr. Cretaceomachilis libanensis, the oldest known bristle-tail of the family Meinertellidae (Machiloidea, Archaeognatha, Insecta) from the Lebanese Amber. Deut. Entomol. Z. 1998, 45, 43–48. [Google Scholar] [CrossRef]
- Grimaldi, D.A.; Engel, M.S.; Nascimbene, P.C. Fossiliferous Cretaceous amber from Myanmar (Burma): Its rediscovery, biotic diversity, and paleontological significance. Am. Mus. Novit. 2002, 2002, 1–71. [Google Scholar] [CrossRef]
- Mendes, L.F.; Wunderlich, J. New data on thysanurans preserved in Burmese amber (Microcoryphia and Zygentoma lnsecta). Soil Org. 2013, 85, 11–22. [Google Scholar]
- Sturm, H.; Poinar, J.G. A new Neomachilellus species from Miocene amber of the Dominican Republic and its phylogenetic relationships (Archaeognatha: Meinertellidae). Entomol. Gen. 1997, 22, 157–170. [Google Scholar] [CrossRef]
- Riquelme, F.; Montejo-Cruz, M.; Luna-Castro, B.; Zuñiga-Mijangos, L. Fossil jumping-bristletail from the Chiapas amber: Neomachilellus (Praeneomachilellus) ezetaelenensis sp. nov.(Microcoryphia: Meinertellidae). Neues Jahrb. Fur Geol. Palaontol.-Abh. 2015, 275, 93–106. [Google Scholar] [CrossRef]
- Montagna, M. Comment on phylogenetic analyses with four new Cretaceous bristletails reveal inter-relationships of Archaeognatha and Gondwana origin of Meinertellidae. Cladistics 2020, 36, 227–231. [Google Scholar] [CrossRef]
- Osozawa, S.; Nel, A. Paleopteran molecular clock: Time drift and recent acceleration. Ecol. Evol. 2024, 14, e70297. [Google Scholar] [CrossRef] [PubMed]
- Selivon, D.; Perondini, A.L.P.; Hernandez-Ortiz, V.; doVal, F.C.; Camacho, A.; Gomes, F.R.; Prezotto, L.F. Genetical, morphological, behavioral, and ecological traits support the existence of three Brazilian species of the Anastrepha fraterculus complex of cryptic species. Front. Ecol. Evol. 2022, 10, 836608. [Google Scholar] [CrossRef]
- Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Zhang, J.Y.; Zhou, C.F.; Gai, Y.H.; Song, D.X.; Zhou, K.Y. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene 2008, 424, 18–24. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Bioinform. Methods Protoc. 1999, 132, 71–91. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Guan, J.Y.; Cao, Y.R.; Dai, X.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Mitogenome analysis of four Lamiinae species (Coleoptera: Cerambycidae) and gene expression responses by Monochamus alternatus when infected with the parasitic nematode, Bursaphelenchus mucronatus. Insects 2021, 12, 453. [Google Scholar] [CrossRef]
- Song, H.; Sheffield, N.C.; Cameron, S.L.; Miller, K.B.; Whiting, M.F. When phylogenetic assumptions are violated: Base compositional heterogeneity and among-site rate variation in beetle mitochondrial phylogenomics. Syst. Entomol. 2010, 35, 429–448. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Illustrator, A. Adobe Illustrator. 2021. Available online: https://adobe.hmzsya.com (accessed on 12 January 2025).
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Cameron, S.L.; Miller, K.B.; D’Haese, C.A.; Whiting, M.F.; Barker, S.C. Mitochondrial genome data alone are not enough to unambiguously resolve the relationships of Entognatha, Insecta and Crustacea sensu lato (Arthropoda). Cladistics 2004, 20, 534–557. [Google Scholar] [CrossRef]
- Carapelli, A.; Liò, P.; Nardi, F.; Van der Wath, E.; Frati, F. Phylogenetic analysis of mitochondrial protein coding genes confirms the reciprocal paraphyly of Hexapoda and Crustacea. BMC Evol. Biol. 2007, 7, S8. [Google Scholar] [CrossRef]
- Podsiadlowski, L. The mitochondrial genome of the bristletail Petrobius brevistylis (Archaeognatha: Machilidae). Insect Mol. Biol. 2006, 15, 253–258. [Google Scholar] [CrossRef]
- Shen, S.Q.; Cai, Y.Y.; Xu, K.K.; Chen, Q.P.; Cao, S.S.; Yu, D.N.; Zhang, J.Y. The complete mitochondrial genome of Pedetontus zhejiangensis (Microcoryphia: Machilidae) and its phylogeny. Mitochondr. DNA B 2020, 5, 3143–3145. [Google Scholar] [CrossRef]
- Guan, J.Y.; Shen, S.Q.; Zhang, Z.Y.; Xu, X.D.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Comparative mitogenomes of two Coreamachilis species (Microcoryphia: Machilidae) along with phylogenetic analyses of Microcoryphia. Insects 2021, 12, 795. [Google Scholar] [CrossRef]
- Cook, C.E.; Yue, Q.; Akam, M. Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc. R. Soc. B Biol. Sci. 2005, 272, 1295–1304. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Schrago, C.G.; Aguiar, B.O.; Mello, B. Comparative evaluation of maximum parsimony and Bayesian phylogenetic reconstruction using empirical morphological data. J. Evol. Biol. 2018, 31, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- FigTree Version 1.4.0. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 January 2025).
- Zhang, J.J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006, 23, 212–226. [Google Scholar] [CrossRef]
- Ho, S.Y.; Phillips, M.J. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst. Biol. 2009, 58, 367–380. [Google Scholar] [CrossRef]
- Püschel, H.P.; O’Reilly, J.E.; Pisani, D.; Donoghue, P.C. The impact of fossil stratigraphic ranges on tip-calibration, and the accuracy and precision of divergence time estimates. Palaeontology 2020, 63, 67–83. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Rasnitsyn, A.P. Taxonomy and morphology of Dasyleptus Brongniart, 1885, with description of a new species (Insecta: Machilida: Dasyleptidae). Russ. Entomol. J. 1999, 8, 145–154. [Google Scholar]
- Keilbach, R. Bibliographie und Liste der Arten tierischer Einschlüsse in fossilen Harzen sowie ihrer Aufbewahrungsorte. Dtsch. Entomol. Z. 1982, 29, 129–286. [Google Scholar] [CrossRef]
- Montagna, M.; Haug, J.T.; Strada, L.; Haug, C.; Felber, M.; Tintori, A. Central nervous system and muscular bundles preserved in a 240 million year old giant bristletail (Archaeognatha: Machilidae). Sci. Rep. 2017, 7, 46016. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Jungreis, I.; Chan, C.S.; Waterhouse, R.M.; Fields, G.; Lin, M.F.; Kellis, M. Evolutionary dynamics of abundant stop codon readthrough. Mol. Biol. Evol. 2016, 33, 3108–3132. [Google Scholar] [CrossRef]
- Bachtrog, D.; Thornton, K.; Clark, A.; Andolfatto, P. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution 2006, 60, 292–302. [Google Scholar]
- Yuan, Y.; Kong, L.; Li, Q. Mitogenome evidence for the existence of cryptic species in Coelomactra antiquata. Genes Genom. 2013, 35, 693–701. [Google Scholar] [CrossRef]
- Powell, J.R.; Moriyama, E.N. Evolution of codon usage bias in Drosophila. Proc. Natl. Acad. Sci. USA 1997, 94, 7784–7790. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.S.; Wu, G.Z.; Wang, Z.F.; Chai, X.W.; Nie, Q.H.; Zhang, X.Q. Mutation bias is the driving force of codon usage in the Gallus gallus genome. DNA Res. 2011, 18, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Mandal, G. Fauna of India Checklist: Arthropoda: Insecta: Archaeognatha. Version 2023, 1, 2. [Google Scholar]
- DiMichele, W.A.; Pfefferkorn, H.W.; Gastaldo, R.A. Response of Late Carboniferous and Early Permian plant communities to climate change. Annu. Rev. Earth Planet. Sci. 2001, 29, 461–487. [Google Scholar] [CrossRef]
- Chaloner, W.G. Spores and land-plant evolution. Rev. Palaeobot. Palynol. 1967, 1, 83–93. [Google Scholar] [CrossRef]
- Cleal, C.J.; Uhl, D.; Cascales-Miñana, B.; Thomas, B.A.; Bashforth, A.R.; King, S.C.; Zodrow, E.L. Plant biodiversity changes in Carboniferous tropical wetlands. Earth-Sci. Rev. 2012, 114, 124–155. [Google Scholar] [CrossRef]
- Lucas, S.G.; DiMichele, W.A.; Opluštil, S.; Wang, X. An Introduction to Ice Ages, Climate Dynamics and Biotic Events: The Late Pennsylvanian World. 2023. Available online: https://www.lyellcollection.org/doi/full/10.1144/SP535-2022-334 (accessed on 1 January 2025).
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef]
- Witts, J.D.; Newton, R.J.; Mills, B.J.; Wignall, P.B.; Bottrell, S.H.; Hall, J.L.; Francis, J.E.; Crame, J.A. The impact of the Cretaceous–Paleogene (K–Pg) mass extinction event on the global sulfur cycle: Evidence from Seymour Island, Antarctica. Geochim. Cosmochim. Acta 2018, 230, 17–45. [Google Scholar] [CrossRef]
- Bateman, R.M. Hunting the Snark: The flawed search for mythical Jurassic angiosperms. J. Exp. Bot. 2020, 71, 22–35. [Google Scholar] [CrossRef]
- Givnish, T.J. Adaptive radiation versus ‘radiation’ and ‘explosive diversification’: Why conceptual distinctions are fundamental to understanding evolution. New Phytol. 2015, 207, 297–303. [Google Scholar] [CrossRef]
- Feldberg, K.; Heinrichs, J.; Schmidt, A.R.; Váňa, J.; Schneider, H. Exploring the impact of fossil constraints on the divergence time estimates of derived liverworts. Plant Syst. Evol. 2013, 299, 585–601. [Google Scholar] [CrossRef]
- Traverse, A. Response of world vegetation to Neogene tectonic and climatic events. Alcheringa 1982, 6, 197–209. [Google Scholar] [CrossRef]
- Laenen, B.; Shaw, B.; Schneider, H.; Goffinet, B.; Paradis, E.; Désamoré, A.; Heinrichs, J.; Villarreal, J.; Gradstein, S.; McDaniel, S. Extant diversity of bryophytes emerged from successive post-Mesozoic diversification bursts. Nat. Commun. 2014, 5, 5134. [Google Scholar] [CrossRef] [PubMed]
- Bippus, A.C.; Flores, J.R.; Hyvönen, J.; Tomescu, A.M. The role of paleontological data in bryophyte systematics. J. Exp. Bot. 2022, 73, 4273–4290. [Google Scholar] [CrossRef]
- Duan, M.; Tu, J.; Lu, Z. Recent advances in detecting mitochondrial DNA heteroplasmic variations. Molecules 2018, 23, 323. [Google Scholar] [CrossRef]
- Ponti, R.; Sannolo, M. The importance of including phenology when modelling species ecological niche. Ecography 2023, 2023, e06143. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Family | Subfamily | Species | Length (bp) | GenBank Accession Number |
|---|---|---|---|---|
| Meinertellidae | / | Nesomachilis australica | 15,474 bp | NC_006895 |
| Machilidae | Petrobiellinae | Petrobiellus sp. 1 JZ-2014 | 15,843 bp | KJ754503 |
| Petrobiellus sp. 2 JZ-2014 | 14,022 bp | KJ754504 | ||
| Machilinae | Allopsontus sp. 1 JZ-2014 | 15,532 bp | KJ754500 | |
| Allopsontus sp. 2 JZ-2014 | 15,538 bp | KJ754501 | ||
| Trigoniophthalmus alternatus | 16,197 bp | NC_010532 | ||
| Machilis hrabei | 15,585 bp | PV126557 | ||
| Coreamachilis coreanus | 15,578 bp | MW752137 | ||
| Coreamachilis songi | 15,570 bp | MW752138 | ||
| Songmachilis xinxiangensis | 15,473 bp | NC_021384 | ||
| Petrobiinae | Petrobius brevistylis | 15,698 bp | NC_007688 | |
| Pedetontinus luanchuanensis | 14,106 bp | KJ754502 | ||
| Pedetontus zhejiangensis | 15,602 bp | NC_051491 | ||
| Pedetontus silvestrii | 15,879 bp | NC_011717 | ||
| Pedetontus silvestrii DD | 15,742 bp | PV126559 | ||
| Pedetontus silvestrii FC | 15,872 bp | PV126560 | ||
| Pedetontus silvestrii GCL | 15,705 bp | PV126561 | ||
| Pedetontus silvestrii TH | 15,770 bp | PV126555 | ||
| Pedetontus silvestrii CD | 15,747 bp | PV126558 | ||
| Pedetontus silvestrii XY | 15,749 bp | PV126556 | ||
| Collembola | Onychiurus orientalis | 12,984 bp | NC_006074 | |
| Podura aquatica | 13,809 bp | NC_006075 | ||
| Best Model | Sites | Partition Names |
|---|---|---|
| GTR + I + G | 1181 | ND3_codon1, ATP6_codon1, COIII_codon1, COII_codon1, Cyt b_codon1 |
| GTR + I + G | 1680 | COI_codon2, ND3_codon2, ATP6_codon2, COII_codon2, COIII_codon2, Cyt b_codon2 |
| TVM + G | 1728 | ND3_codon3, COII_codon3, ATP8_codon3, ATP6_codon3, COI_codon3, COIII_codon3, Cyt b_codon3 |
| GTR + I + G | 528 | ATP8_codon2, ATP8_codon1, ND2_codon1, ND6_codon1 |
| SYM + I + G | 499 | COI_codon1 |
| GTR + I + G | 1327 | ND1_codon1, ND5_codon1, ND4L_codon1, ND4_codon1 |
| GTR + I + G | 1327 | ND4_codon2, ND5_codon2, ND4L_codon2, ND1_codon2 |
| HKY + G | 1327 | ND4L_codon3, ND5_codon3, ND4_codon3, ND1_codon3 |
| TVM + G | 432 | ND2_codon2, ND6_codon2 |
| HKY + G | 432 | ND2_codon3, ND6_codon3 |
| CD | DD | FC | XY | GCL | TH | |
|---|---|---|---|---|---|---|
| P. silvestrii CD | ||||||
| P. silvestrii DD | 0.1827 | |||||
| P. silvestrii FC | 0.1794 | 0.0164 | ||||
| P. silvestrii XY | 0.1871 | 0.0674 | 0.0672 | |||
| P. silvestrii GCL | 0.1808 | 0.1721 | 0.1710 | 0.1170 | ||
| P. silvestrii TH | 0.1941 | 0.1178 | 0.1148 | 0.1165 | 0.1783 | |
| P. silvestrii NC_011717 | 0.1069 | 0.1848 | 0.1822 | 0.1916 | 0.1848 | 0.1954 |
| Nodes/Clades | Mean Divergence Time (Mya) | 95% HPD Range (Mya) |
|---|---|---|
| Meinertellidae | 127.76 | 125.45, 130.00 |
| Machilidae | 238.46 | 235.01, 241.99 |
| Meinertellidae & Machilidae | 301.19 | 298.88, 303.68 |
| Meinertellidae & Petrobiellinae | 48.24 | 15.89, 101.83 |
| Machilinae & Petrobiinae | 153.99 | 89.91, 227.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cen, W.; Li, J.-W.; He, J.-T.; Chen, X.-Y.; Li, L.-Y.; Storey, K.B.; Yu, D.-N.; Zhang, J.-Y. Morpho-Molecular Discordance and Cryptic Diversity in Jumping Bristletails: A Mitogenomic Analysis of Pedetontus silvestrii (Insecta: Archaeognatha: Machilidae). Insects 2025, 16, 452. https://doi.org/10.3390/insects16050452
Cen W, Li J-W, He J-T, Chen X-Y, Li L-Y, Storey KB, Yu D-N, Zhang J-Y. Morpho-Molecular Discordance and Cryptic Diversity in Jumping Bristletails: A Mitogenomic Analysis of Pedetontus silvestrii (Insecta: Archaeognatha: Machilidae). Insects. 2025; 16(5):452. https://doi.org/10.3390/insects16050452
Chicago/Turabian StyleCen, Wei, Jia-Wen Li, Jia-Tao He, Xin-Yu Chen, Luo-Ying Li, Kenneth B. Storey, Dan-Na Yu, and Jia-Yong Zhang. 2025. "Morpho-Molecular Discordance and Cryptic Diversity in Jumping Bristletails: A Mitogenomic Analysis of Pedetontus silvestrii (Insecta: Archaeognatha: Machilidae)" Insects 16, no. 5: 452. https://doi.org/10.3390/insects16050452
APA StyleCen, W., Li, J.-W., He, J.-T., Chen, X.-Y., Li, L.-Y., Storey, K. B., Yu, D.-N., & Zhang, J.-Y. (2025). Morpho-Molecular Discordance and Cryptic Diversity in Jumping Bristletails: A Mitogenomic Analysis of Pedetontus silvestrii (Insecta: Archaeognatha: Machilidae). Insects, 16(5), 452. https://doi.org/10.3390/insects16050452