


The Characteristics and Functions of SSRs and SNPs Based on the Transcriptome of Tuta absoluta Exposed to Different Concentrations of Abamectin and Chlorantraniliprole

Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Insects
2.2. Sample Processing and RNA Isolation
2.3. Sequencing and Data Analysis
3. Results
3.1. SSR Distribution of Transcriptome
3.2. Analysis of Different Repeat Types of SSR Loci
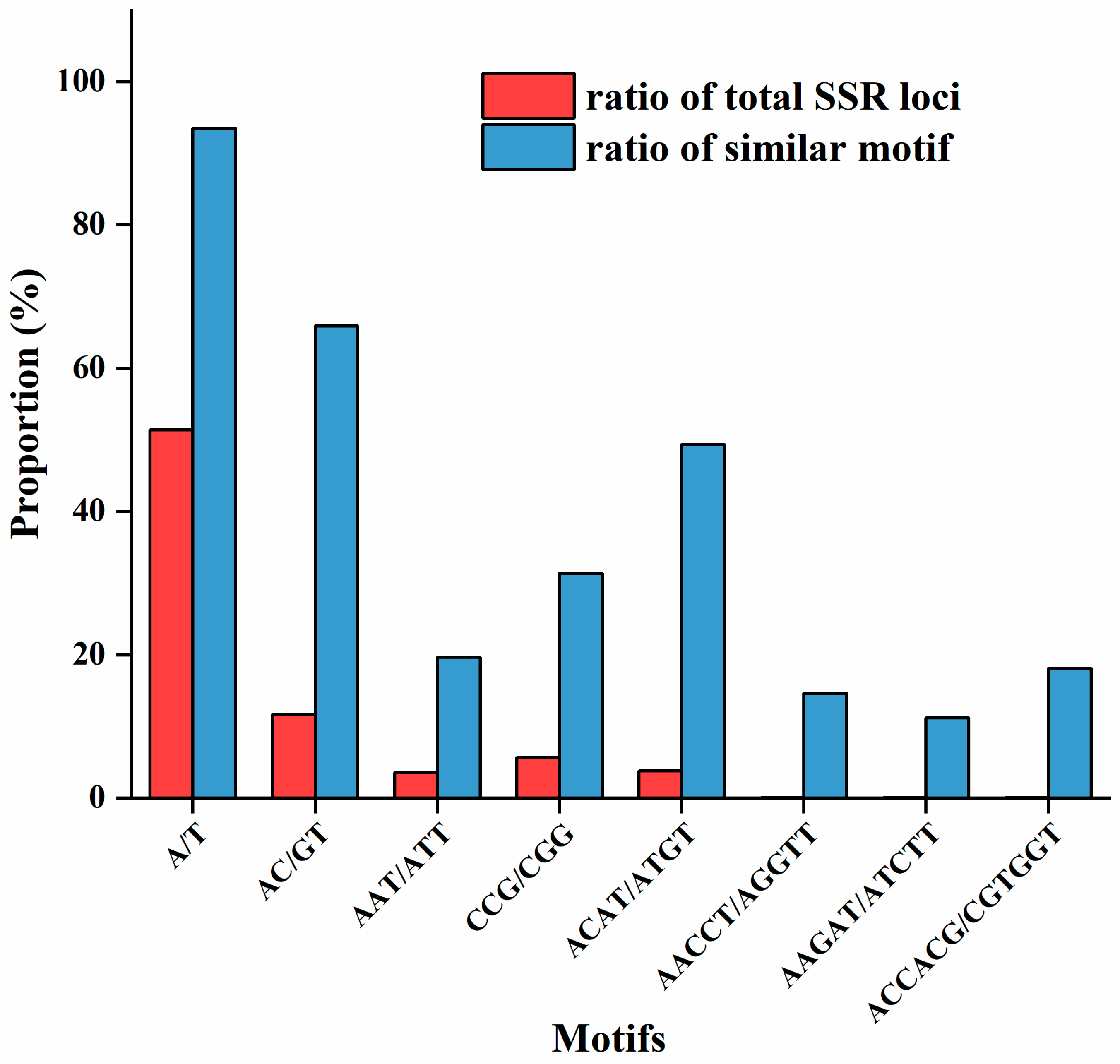
3.3. Analysis of SSR Motif Types and Repeat Times
3.4. Distribution Characteristics of SSR Motif Length
3.5. GO Functional Classification of of SSR-Transcripts
3.6. COG Pathway Analysis of SSR-Transcripts
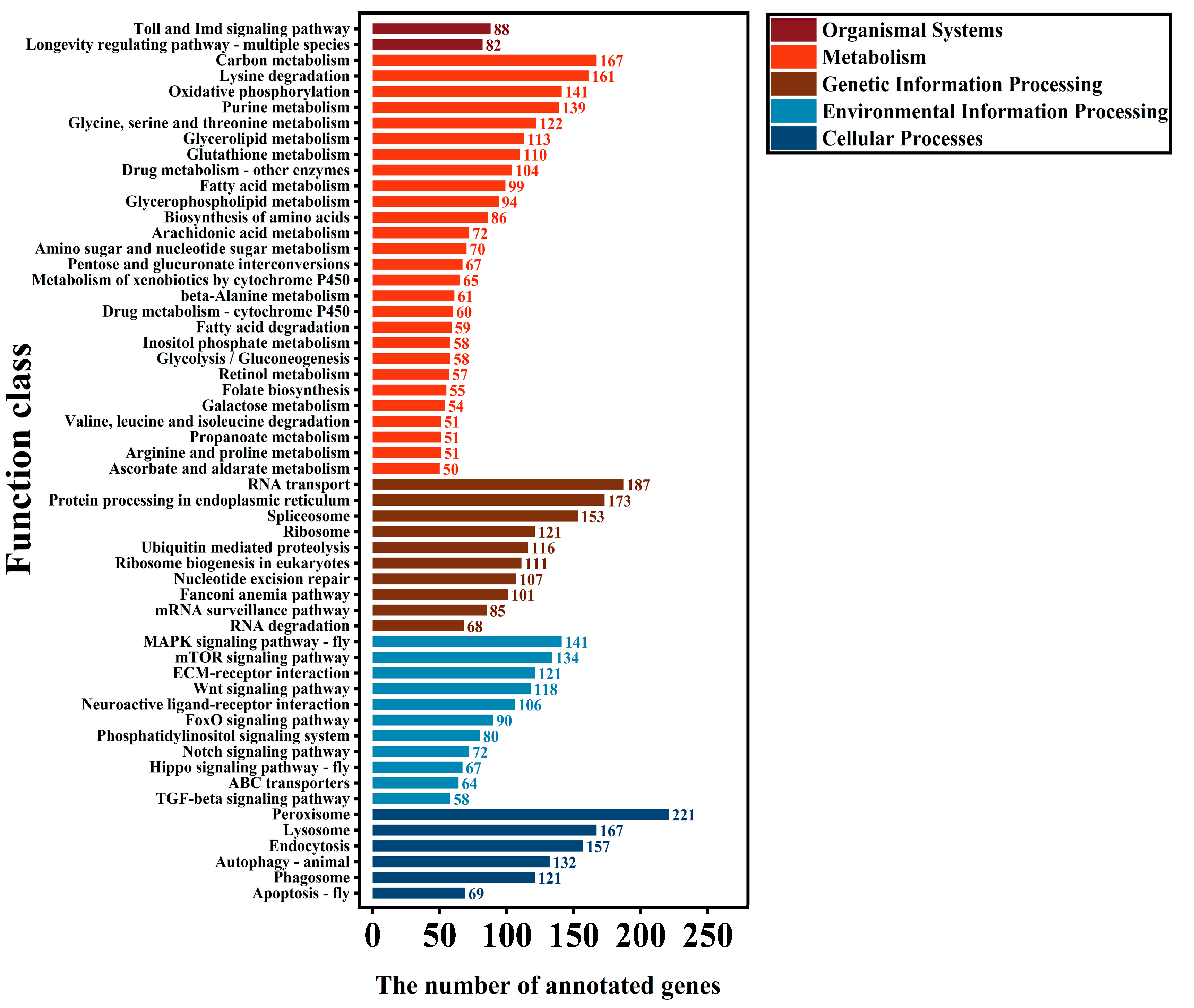
3.7. KEGG Metabolic Pathway Analysis of SSR-Transcripts
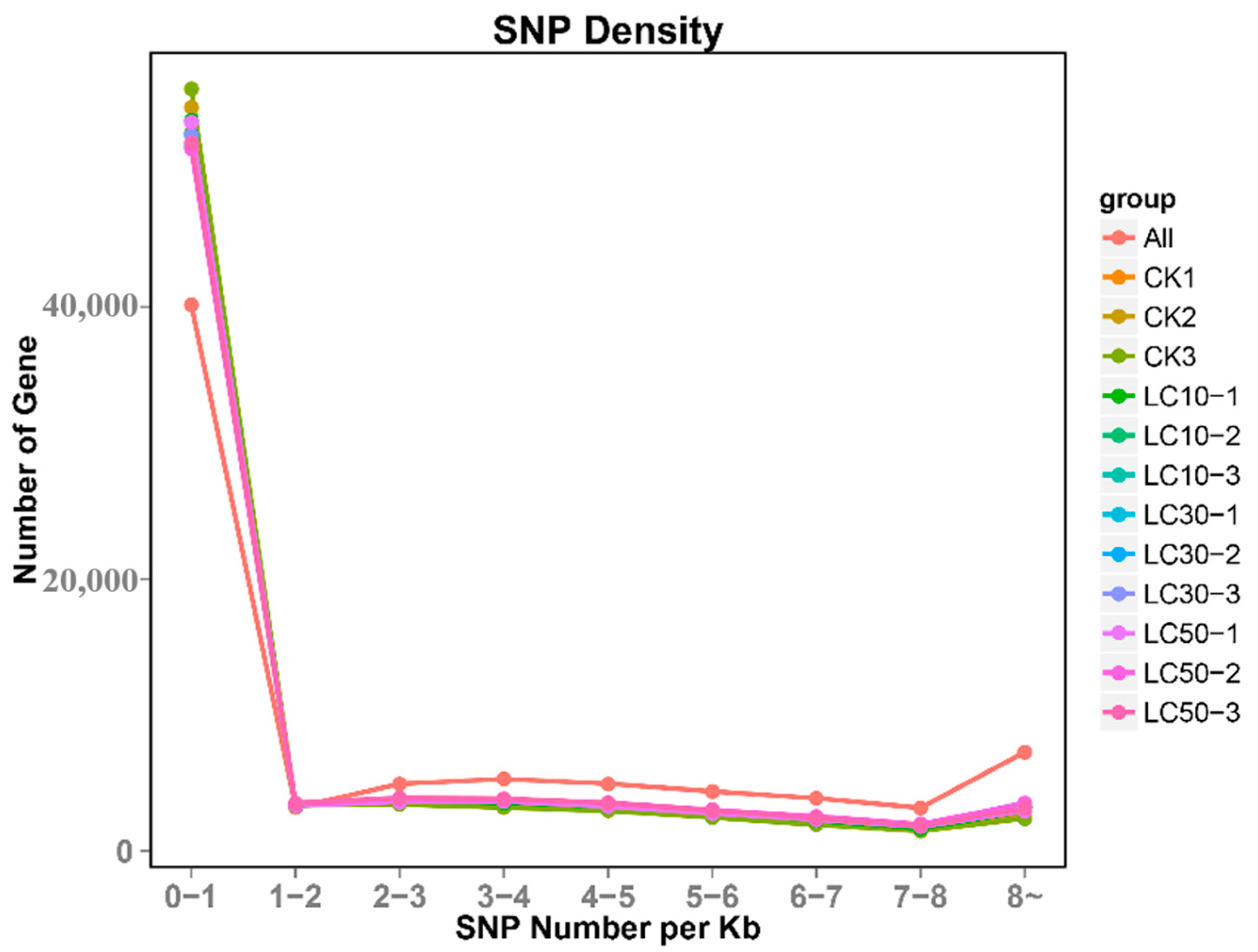
3.8. SNP Loci Analysis
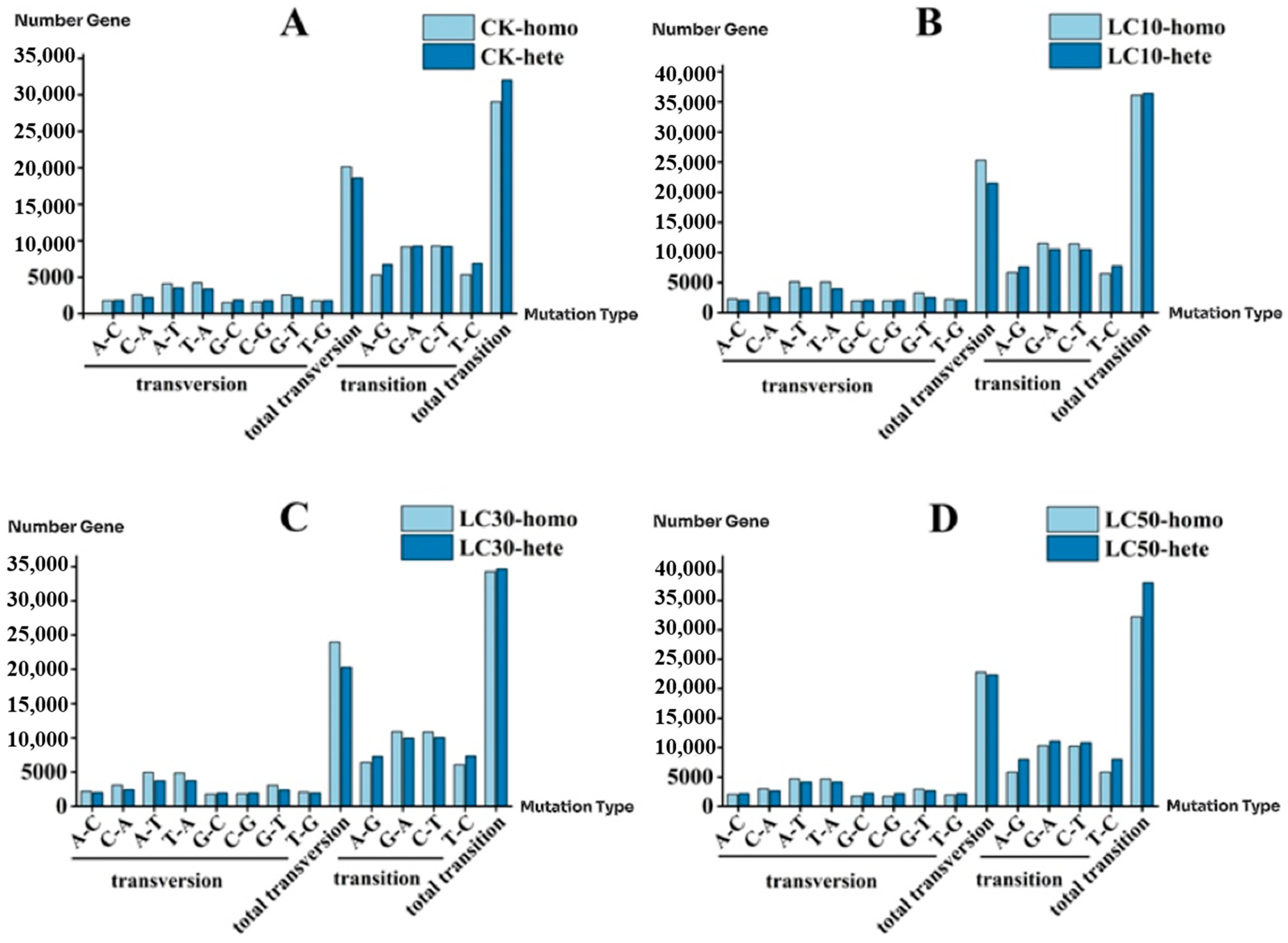
3.9. SNP Transitions and Transversions
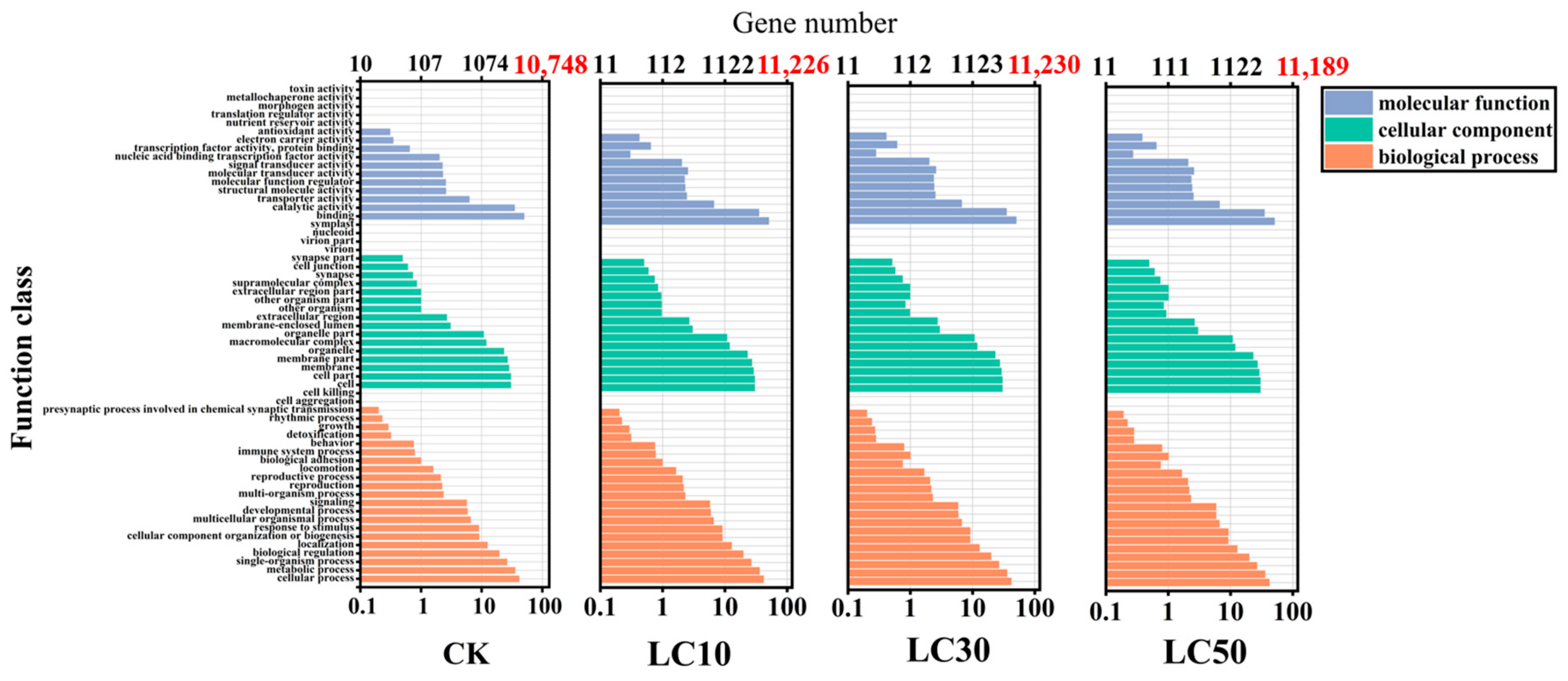
3.10. GO Functional Classification of SNP-Transcripts
3.11. COG Pathway Analysis of SNP-Transcripts
3.12. KEGG Metabolic Pathway Analysis of SNP-Transcripts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campos, M.R.; Biondi, A.; Adiga, A.; Guedes, R.N.C.; Desneux, N. From the Western Palaearctic Region to beyond: Tuta Absoluta 10 Years after Invading Europe. J. Pest. Sci. 2017, 90, 787–796. [Google Scholar] [CrossRef]
- Xian, X.; Han, P.; Wang, S.; Zhang, G.; Liu, W.; Desneux, N.; Wan, F. The Potential Invasion Risk and Preventive Measures against the Tomato Leafminer Tuta Absoluta in China. Entomol. Gen. 2017, 36, 319–333. [Google Scholar] [CrossRef]
- Zhang, G.F.; Ma, D.Y.; Wang, Y.S.; Gao, Y.H.; Liu, W.X.; Zhang, R.; Fu, W.J.; Xian, X.Q.; Wang, J.; Kang, M.; et al. First report of the South American tomato leafminer, Tuta absoluta (Meyrick), in China. J. Integr. Agric. 2020, 19, 1912–1917. [Google Scholar] [CrossRef]
- Zhang, Z.K.; Wu, S.Y.; Lei, Z.R. Occurrence and control of Tuta absoluta, a new agricultural invasive organism in Ningxia. Chin. Melon Veg. 2022, 35, 111–116. [Google Scholar]
- Campos, M.R.; Amiens-Desneux, E.; Béarez, P.; Soares, M.A.; Ponti, L.; Biondi, A.; Harwood, J.D.; Desneux, N. Impact of Low Temperature and Host Plant on Tuta absoluta. Entomol. Exp. Et. Appl. 2021, 169, 984–996. [Google Scholar] [CrossRef]
- Zhang, G.; Xian, X.; Zhang, Y.; Liu, W.; Liu, H.; Feng, X.; Ma, D.; Wang, Y.; Gao, Y.; Zhang, R.; et al. Outbreak of the South American Tomato Leafminer, Tuta Absoluta, in the Chinese Mainland: Geographic and Potential Host Range Expansion. Pest. Manag. Sci. 2021, 77, 5475–5488. [Google Scholar] [CrossRef]
- Silva, G.A.; Queiroz, E.A.; Arcanjo, L.P.; Lopes, M.C.; Araújo, T.A.; Galdino, T.S.V.; Samuels, R.I.; Rodrigues-Silva, N.; Picanço, M.C. Biological Performance and Oviposition Preference of Tomato Pinworm Tuta Absoluta When Offered a Range of Solanaceous Host Plants. Sci. Rep. 2021, 11, 1153. [Google Scholar] [CrossRef]
- Medeiros, M.A.D.; Boas, G.L.V.; Vilela, N.J.; Carrijo, O.A. A preliminar survey on the biological control of South American tomato pinworm with the parasitoid Trichogramma pretiosum in greenhouse models. Hortic. Bras. 2009, 27, 80–85. [Google Scholar] [CrossRef]
- Cherif, A.; Attia-Barhoumi, S.; Mansour, R.; Zappala, L.; Grissa-Lebdi, K. Elucidating key biological parameters of Tuta absoluta on different host plants and under various temperature and relative humidity regimes. Entomol. Gen. 2019, 39, 1–7. [Google Scholar] [CrossRef]
- Barati, R.; Hejazi, M.J.; Mohammadi, S.A. Insecticide susceptibility in Tuta absoluta (Lepidoptera: Gelechiidae) and metabolic characterization of resistance to diazinon. J. Econ. Entomol. 2018, 111, 1551–1557. [Google Scholar] [CrossRef]
- Martins, M.R.; Nascimento, A.F.S.; de Sena Fernandes, M.E.; Tronto, J.; da Fonseca, L.F.; Fernandes, F.L. Chlorantraniliprole mediating the survival and behavior of adults of Tuta absoluta(meyrick) (lepidoptera: Gelechiidae) in tomato. Int. J. Trop. Insect Sci. 2024, 44, 2879–2888. [Google Scholar] [CrossRef]
- Yao, J.; Qu, C.; Li, Y.; Wang, Y.; Luo, C.; Guedes, R.N.C.; Wang, R. Sublethal and transgenerational effects of tetraniliprole on the tomato pinworm Phthorimaea (=Tuta) absoluta (Lepidoptera: Gelechiidae). Crop Prot. 2025, 187, 106953. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Zhang, Y.; Sun, Z.; Li, Y.; Ding, J.; Zhang, G.; Du, E.; Zi, X.; Tian, C.; et al. Role of Enterococcus mundtii in gut of the tomato leaf miner (Tuta absoluta) to detoxification of Chlorantraniliprole. Pestic. Biochem. Physiol. 2024, 204, 106060. [Google Scholar] [CrossRef] [PubMed]
- Roditakis, E.; Mavridis, K.; Riga, M.; Vasakis, E.; Morou, E.; Rison, J.L.; Vontas, J. Identification and detection of indoxacarb resistance mutations in the para sodium channel of the tomato leafminer, Tuta absoluta. Pest. Manag. Sci. 2017, 73, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Prasannakumar, N.R.; Jyothi, N.; Prasadbabu, K.; Ramkumar, G.; Asokan, R.; Saroja, S.; Sridhar, V. Evidence-based insecticide resistance in South American tomato leaf miner, Phthorimaea absoluta (Meyrick) under laboratory selection. Bull. Entomol. Res. 2023, 113, 419–429. [Google Scholar] [CrossRef]
- Guedes, R.N.C.; Roditakis, E.; Campos, M.R.; Haddi, K.; Bielza, P.; Siqueira, H.A.; Tsagkarakou, A.; Vontas, J.; Nauen, R. Insecticide resistance in the tomato pinworm Tuta absoluta: Patterns, spread, mechanisms, management and outlook. J. Pest. Sci. 2019, 92, 1329–1342. [Google Scholar] [CrossRef]
- Mashtoly, T.A.; El-Beltagi, H.S.; Almujam, A.N.; Othman, M.N. The potential of a novel concept of an integrated bio and chemical formulate based on an entomopathogenic bacteria, Bacillus thuringiensis, and a chemical insecticide to control tomato leafminer, Tuta absoluta ‘(Meyrick)’(Lepidoptera: Gelechiidae). Sustainability 2022, 14, 10582. [Google Scholar] [CrossRef]
- Liu, M.; Xiao, F.; Zhu, Y.; Fu, D.; Wang, Z.; Xiao, R. Combined PacBio Iso-Seq and Illumina RNA-Seq analysis of the Tuta absoluta (Meyrick) transcriptome and cytochrome P450 genes. Insects 2023, 14, 363. [Google Scholar] [CrossRef]
- Stolle, E.; Kidner, J.H.; Moritz, R.F. Patterns of evolutionary conservation of microsatellites (SSRs) suggest a faster rate of genome evolution in Hymenoptera than in Diptera. Genome Biol. Evol. 2013, 5, 151–162. [Google Scholar] [CrossRef]
- Buschiazzo, E.; Gemmell, N.J. The Rise, Fall and Renaissance of Microsatellites in Eukaryotic Genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple Sequences with Complex Evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, M.R.; Kumar, P.; Singh, R.P.; Shanker, R.; Singh, J.; Bharti, M.K.; Singh, R.; Verma, H.; Gangwar, L.K.; Gaurav, S.S.; et al. De novo transcriptome assembly, annotation and SSR mining data of Hellula undalis (Fabr.) (Lepidoptera: Pyralidae), the cabbage webworm. J. Genet. Eng. Biotechnol. 2024, 22, 100393. [Google Scholar] [CrossRef] [PubMed]
- Kattupalli, D.; Barbadikar, K.M.; Balija, V.; Ballichatla, S.; Padmakumari, A.P.; Saxena, S.; Gaikwad, K.; Yerram, S.; Kokku, P.; Madhav, M.S. The Draft Genome of Yellow Stem Borer, an Agriculturally Important Pest, Provides Molecular Insights into Its Biology, Development and Specificity Towards Rice for Infestation. Insects 2021, 12, 563. [Google Scholar] [CrossRef]
- Miao, X.X.; Xub, S.J.; Li, M.H.; Li, M.W.; Huang, J.H.; Dai, F.Y.; Marino, S.W.; Mills, D.R.; Zeng, P.; Mita, K.; et al. Simple sequence repeat-based consensus linkage map of Bombyx mori. Proc. Natl. Acad. Sci. USA 2005, 102, 16303–16308. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Zhao, Y.; Xu, L.; Yi, H.; Zhang, Y.; Jia, X.; Zhao, H.; Zhao, J.; Wang, F.; Wang, F. An integrated strategy for target SSR genoty** with toleration of nucleotide variations in the SSRs and flanking regions. BMC Bioinform. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Moghaieb, R.E.; Khashaba, E.H.; Abd El Azim, A.M.; Ibrahim, S.A. Genetic diversity studies and screening for rice stem borer (Chilo Agamemnon) resistance in six Egyptian rice cultivars (Oryza sativa L.) using DNA based-markers. Genet. Resour. Crop Evol. 2021, 68, 2313–2323. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Q.; Li, J.; Zeng, C.; Hu, S.; Yu, J. The study of neighboring nucleotide composition and transition/transversion bias. Sci. China Ser. C. 2006, 49, 395–402. [Google Scholar] [CrossRef]
- Lijavetzky, D.; Cabezas, J.A.; Ibáñez, A.; Rodríguez, V.; Martínez-Zapater, J.M. High throughput snp discovery and genotyping in grapevine (vitis vinifera l.) by combining a re-sequencing approach and snplex technology. BMC Genom. 2007, 8, 424. [Google Scholar] [CrossRef]
- Zhu, G.; Zhong, D.; Cao, J.; Zhou, H.; Li, J.; Liu, Y.; Bai, L.; Xu, S.; Wang, M.G.; Zhou, G.; et al. Transcriptome profiling of pyrethroid resistant and susceptible mosquitoes in the malaria vector, anopheles sinensis. BMC Genom. 2014, 15, 448. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Bonizzoni, M.; Zhong, D.; Li, Y.; Zhou, G.; Nguyen, H.; Tong, S.; Yan, G.; Chen, X.G. Comparative transcriptome analysis and RNA interference reveal CYP6A8 and SNPs related to pyrethroid resistance in Aedes albopictus. PLoS Neglected Trop. Dis. 2018, 12, e0006828. [Google Scholar] [CrossRef]
- Kassa, M.T.; Haas, S.; Schliephake, E.; Lewis, C.; You, F.M.; Pozniak, C.J.; Krämer, I.; Perovic, D.; Sharpe, D.G.; Fobert, P.R.; et al. A saturated snp linkage map for the orange wheat blossom midge resistance gene sm1. Theor. Appl. Genet. 2016, 129, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.X.; Hartman, G.L. Characterization of insect resistance loci in the USDA soybean germplasm collection using genome-wide association studies. Front. Plant Sci. 2017, 8, 670. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, F.; Chiesa, O.; Panini, M.; Massimino Cocuzza, G.E.; Mazzoni, E. Survey of target site mutations linked with insecticide resistance in Italian populations of Aphis gossypii. Pest. Manag. Sci. 2024, 80, 4361–4370. [Google Scholar] [CrossRef]
- Puggioni, V.; Chiesa, O.; Panini, M.; Mazzoni, E. Qualitative Sybr Green real-time detection of single nucleotide polymorphisms responsible for target-site resistance in insect pests: The example of Myzus persicae and Musca domestica. Bull. Entomol. Res. 2017, 107, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Lin, A.; Ji, M.; Jing, S.; Song, Z.; Zhenxaing Li, Z.; Song, L.; Sun, J.; Cheng, Q. Comprehensive sex- and tissue-specific transcriptome of the destructive invasive pest Tuta absoluta adults. Sci. Data 2024, 11, 1429. [Google Scholar] [CrossRef]
- Bali, G.K.; Singh, S.K.; Chauhan, V.K.; Joshi, N.; Bhat, F.A.; Malla, W.A.; Ramanujam, B.; Varshney, R.; Kour, M.; Pandit, R.S. An insight in proteome profiling of Tuta absoluta larvae after entomopathogenic fungal infection. Sci. Data 2022, 9, 507. [Google Scholar] [CrossRef]
- Huang, C.; Ou, X.; Wang, Y.; Zhou, Y.; Zhang, G.; Liu, W.; Wan, F.; Jiang, H.; Zhang, Y. Genome-Wide Identification, Evolution, and Female-Biased Expression Analysis of Odorant Receptors in Tuta absoluta (Lepidoptera: Gelechiidae). Life 2024, 14, 872. [Google Scholar] [CrossRef]
- Lewald, K.M.; Tabuloc, C.A.; Godfrey, K.E.; Arnó, J.; Perini, C.R.; Guedes, J.C.; Chiu, J.C. Genome assembly and population sequencing reveal three populations and signatures of insecticide resistance of Tuta absoluta in Latin America. Genome Biol. Evol. 2023, 15, evad060. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Li, F.; Wang, R.; Qu, C.; Fu, N.; Luo, C.; Xu, Y. Sequencing and Characterization of the Invasive Sycamore Lace Bug Corythucha Ciliata (Hemiptera: Tingidae) Transcriptome. PLoS ONE 2016, 11, e0160609. [Google Scholar] [CrossRef]
- Ouyang, H.; Wang, X.; Zheng, X.; Lu, W.; Qin, F.; Chen, C. Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera Dorsalis (Hendel). Insects 2021, 12, 938. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, W.; Zhou, Y.; Wu, J.; Zhou, X. Transcriptome and Comparative Gene Expression Analysis of Sogatella Furcifera (Horváth) in Response to Southern Rice Black-Streaked Dwarf Virus. PLoS ONE 2012, 7, e36238. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Li, Y.H.; Yang, S.; Li, Q.W. De Novo Assembly and Characterization of the Global Transcriptome for Rhyacionia Leptotubula Using Illumina Paired-End Sequencing. PLoS ONE 2013, 8, e81096. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.M.; Tao, J.; Zong, S.X. Feature analysis of simple sequence repeats in Eogystia hippophaecolus transcriptome. Environ. Entomol. 2017, 39, 605–610. [Google Scholar]
- Deng, K.; Deng, R.; Fan, J.; Chen, E. Transcriptome Analysis and Development of Simple Sequence Repeat (SSR) Markers in Zingiber Striolatum Diels. Physiol. Mol. Biol. Plants 2018, 24, 125–134. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Zhang, X.; Hou, Y.; Wang, G. Development of Microsatellite Markers for the Nipa Palm Hispid Beetle, Octodonta Nipae (Maulik). Can. J. Infect. Dis. Med. Microbiol. 2018, 2018, 9139306. [Google Scholar] [CrossRef]
- Duan, X.; Wang, K.; Su, S.; Tian, R.; Li, Y.; Chen, M. De Novo Transcriptome Analysis and Microsatellite Marker Development for Population Genetic Study of a Serious Insect Pest, Rhopalosiphum Padi (L.) (Hemiptera: Aphididae). PLoS ONE 2017, 12, e0172513. [Google Scholar] [CrossRef]
- Hu, Y.H.; Li, M.; Zhang, H.F.; Li, S.C.; Wang, Q.; Zhao, H.L. The Information Analysis of SSR Loci in the Mythimna separate (Walker) Transcriptome. J. Shanxi Agric. Univ. (Nat. Sci. Ed.) 2015, 35, 484–489. [Google Scholar]
- Fu, S.; Duan, Y.; Wang, S.; Ren, Y.; Bu, W. Comparative Transcriptomic Analysis of Riptortus Pedestris (Hemiptera: Alydidae) to Characterize Wing Formation across All Developmental Stages. Insects 2021, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Mo, S.H.; Liu, Y.; Wei, D. Transcriptomic profiling of various developmental stages of Aphis Aurantii to provide a genetic resource for gene expression and SSR analysis. Front. Physiol. Front. Genet. 2020, 11, 578939. [Google Scholar] [CrossRef] [PubMed]
- Meglécz, E.; Nève, G.; Biffin, E.; Gardner, M.G. Breakdown of Phylogenetic Signal: A Survey of Microsatellite Densities in 454 Shotgun Sequences from 154 Non Model Eukaryote Species. PLoS ONE 2012, 7, e40861. [Google Scholar] [CrossRef]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and Experimental Analysis of Microsatellites in Rice (Oryza sativa, L.): Frequency, Length Variation, Transposon Associations, and Genetic Marker Potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Moe, K.T.; Chung, J.W.; Cho, Y.I.; Moon, J.K.; Ku, J.H.; Jung, J.K.; Lee, J.; Park, Y.J. Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of mungbean. J. Integr. Plant Biol. 2011, 53, 63–73. [Google Scholar] [CrossRef]
- Somers, D.J.; Kirkpatrick, R.; Moniwa, M.; Walsh, A. Mining single-nucleotide polymorphisms from hexaploid wheat ESTs. Genome 2003, 46, 431–437. [Google Scholar] [CrossRef]
- Wang, X.; Sun, L.Y.; Liu, J.; Xiao, R. Mining and functional analyzing of SNP related to temperature stress in transcriptome of Pardosa pseudoannulata. Acta Agric. Boreali-Sin. 2019, 34, 331–339. [Google Scholar]
- Gao, Z.; Luo, W.; Liu, H.; Zeng, C.; Liu, X.; Yi, S.; Wang, W. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar] [CrossRef]
- Liu, H.; Li, Z.; Li, F.; Lv, Z.; Wu, S.; Tan, W. SNP and InDel Analysis Based on Tetrastichus brontispae Transcriptome. Chin. J. Trop. Crops 2021, 42, 2828–2833. [Google Scholar]
- Varshney, R.K.; Beier, U.; Khlestkina, E.K.; Kota, R.; Korzun, V.; Graner, A.; Borner, A. Single nucleotide polymorphisms in rye (Secale cereale L.): Discovery, frequency, and applications for genome mapping and diversity studies. Theor. Appl. Genet. 2007, 114, 1105–1116. [Google Scholar] [CrossRef]
- Chang, H.; Gao, Y.; Wang, S.; Wang, X.; Liu, Y.; Sun, H. Analysis of SSR and SNP Loci in Agrotis segetum Based on Transcriptome Sequencing. Guangdong Agric. Sci. 2022, 49, 118–125. [Google Scholar]
- Zhang, X.; Wan, G.; Li, Z.; Wu, Q.; Xiong, S.; Wang, X.; You, L.; Hu, Y. Integrated transcriptome and SNP analysis reveals sex-related genetic insights in the black-spotted frog (Pelophylax nigromaculatus). Aquac. Int. 2024, 32, 6227–6249. [Google Scholar] [CrossRef]
- Raizada, A.; Souframanien, J. Transcriptome sequencing, de novo assembly, characterisation of wild accession of blackgram (Vigna mungo var. silvestris) as a rich resource for development of molecular markers and validation of SNPs by high resolution melting (HRM) analysis. BMC Plant Biol. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Yang, L.; Wang, W.; Huang, J.; Zhang, J.; Wang, Z.C. Identification and functional analysis of snp from transcriptome of cobia (Rachycentron canadum) in response to hypoxia stress. Acta Oceanol. Sin. 2022, 44, 113–124. [Google Scholar]
- Roditakis, E.; Steinbach, D.; Moritz, G.; Vasakis, E.; Stavrakaki, M.; Ilias, A.; García-Vidal, L.; Martínez-Aguirre, M.R.; Bielza, P.; Morou, E.; et al. Ryanodine receptor point mutations confer diamide insecticide resistance in tomato leafminer, Tuta absoluta (Lepidoptera: Gelechiidae). Insect Biochem. Mol. Biol. 2017, 80, 11–20. [Google Scholar] [CrossRef]
- Grant, C.; Singh, K.S.; Hayward, A.; Hunt, B.J.; Troczka, B.J.; Pym, A.; Ahn, S.J.; Zeng, B.; Gao, C.F.; Leroux, A.; et al. Overexpression of the UDP-glycosyltransferase UGT34A23 confers resistance to the diamide insecticide chlorantraniliprole in the tomato leafminer, Tuta absoluta. Insect Biochem. Mol. Biol. 2023, 159, 1039. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsatellite or SSR Distribution | Data |
|---|---|
| Total number of sequences examined | 60,386 |
| Total size of examined sequences (bp) | 164,477,748 |
| Total number of identified SSRs | 25,123 |
| Occurrence frequency (%) | 28.62 |
| Appearance frequency (%) | 41.6 |
| Number of SSR containing sequences | 17,284 |
| Number of sequences containing more than 1 SSR | 4824 |
| Number of SSRs present in compound formation | 2028 |
| Unit Size | Number of SSRs | Proportion (%) | Frequency (%) |
|---|---|---|---|
| mononucleotide | 13,810 | 54.97 | 22.87 |
| dinucleotide | 4475 | 17.81 | 7.41 |
| trinucleotide | 4567 | 18.18 | 7.56 |
| tetranucleotide | 1944 | 7.74 | 3.22 |
| pentanucleotide | 178 | 0.71 | 0.29 |
| hexanucleotide | 149 | 0.59 | 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Mu, M.; Wang, Z.; Zhang, H.; Song, Y.; Xiao, R. The Characteristics and Functions of SSRs and SNPs Based on the Transcriptome of Tuta absoluta Exposed to Different Concentrations of Abamectin and Chlorantraniliprole. Insects 2025, 16, 446. https://doi.org/10.3390/insects16050446
Zhang F, Mu M, Wang Z, Zhang H, Song Y, Xiao R. The Characteristics and Functions of SSRs and SNPs Based on the Transcriptome of Tuta absoluta Exposed to Different Concentrations of Abamectin and Chlorantraniliprole. Insects. 2025; 16(5):446. https://doi.org/10.3390/insects16050446
Chicago/Turabian StyleZhang, Fanxue, Meimei Mu, Zonglin Wang, Haoran Zhang, Yilan Song, and Rong Xiao. 2025. "The Characteristics and Functions of SSRs and SNPs Based on the Transcriptome of Tuta absoluta Exposed to Different Concentrations of Abamectin and Chlorantraniliprole" Insects 16, no. 5: 446. https://doi.org/10.3390/insects16050446
APA StyleZhang, F., Mu, M., Wang, Z., Zhang, H., Song, Y., & Xiao, R. (2025). The Characteristics and Functions of SSRs and SNPs Based on the Transcriptome of Tuta absoluta Exposed to Different Concentrations of Abamectin and Chlorantraniliprole. Insects, 16(5), 446. https://doi.org/10.3390/insects16050446