
Impact of Ectropis grisescens Warren (Lepidoptera: Geometridae) Infestation on the Tea Plant Rhizosphere Microbiome and Its Potential for Enhanced Biocontrol and Plant Health Management

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tea Plant Cultivation
2.2. Insects Rearing
2.3. Insect Infestation
2.4. Soil Samples 16S rRNA Gene Amplicon Sequence
2.5. Plant Samples RNA-Seq, De Novo Assembly, and Functional Annotation
2.6. Plant Samples Identification of Differentially Expressed Genes (DEGs)
2.7. Co-Occurrence Network Analyses
2.8. Metagenomic Sequencing
2.9. Isolation and Characterization of Burkholderia
2.10. Effects of Burkholderia Isolations on Plant Growth
2.11. Larvae Feeding Selection Bioassay
2.12. Chemical Defense Substances Analysis
2.13. Statistical Analysis
3. Results
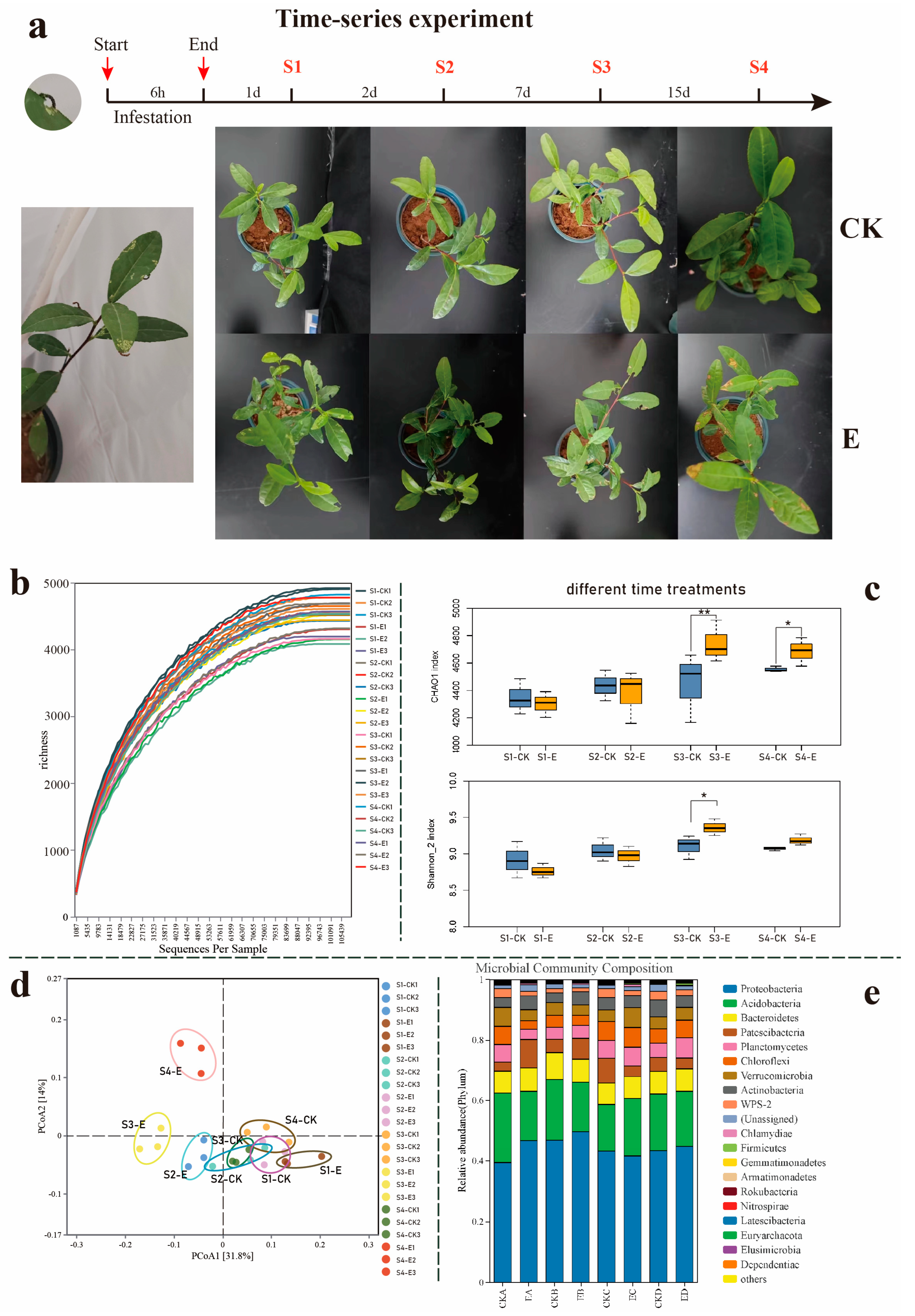
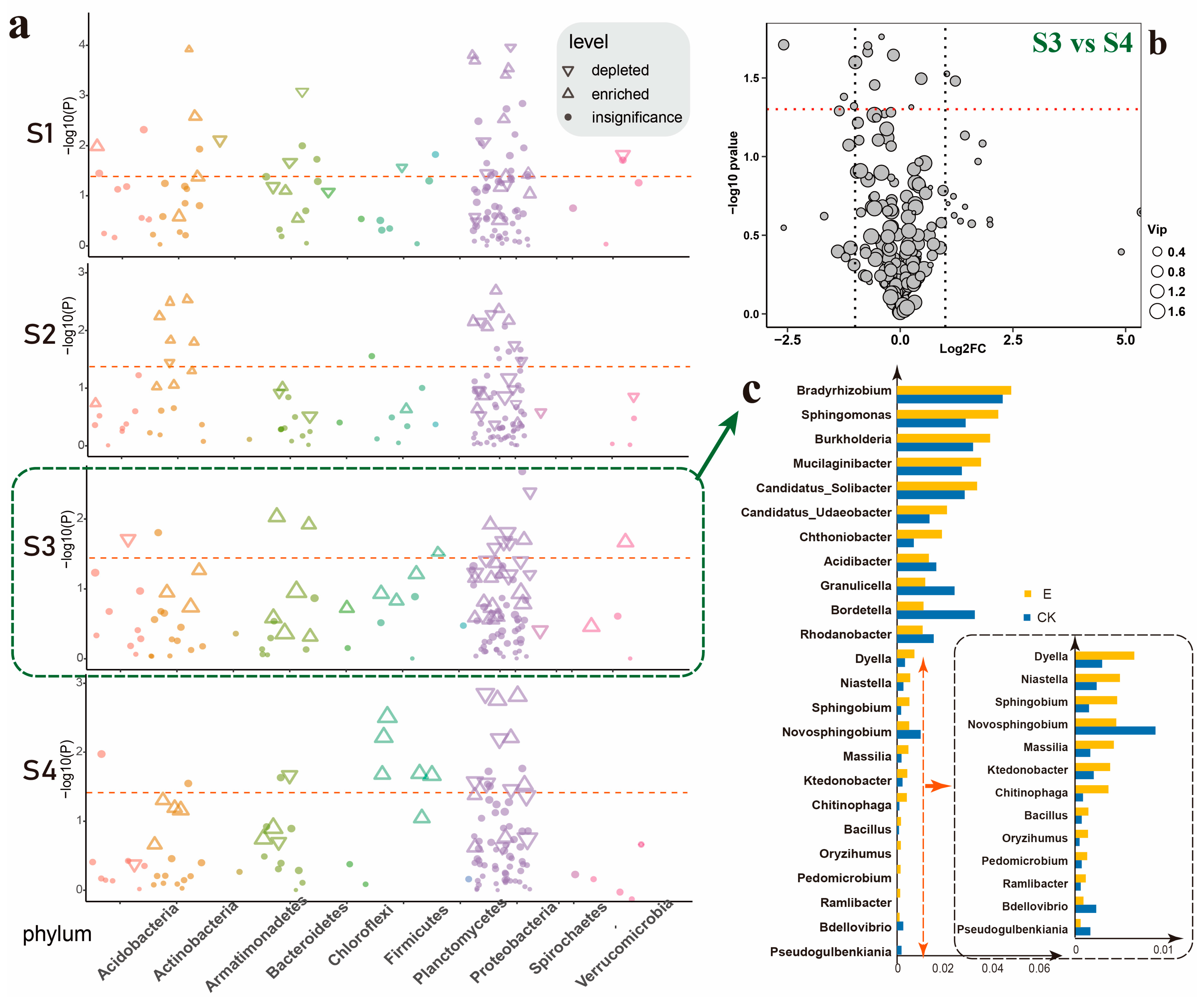
3.1. Ectropis grisescens Infestation Restructures the Tea Rhizosphere Microbiome Assembly
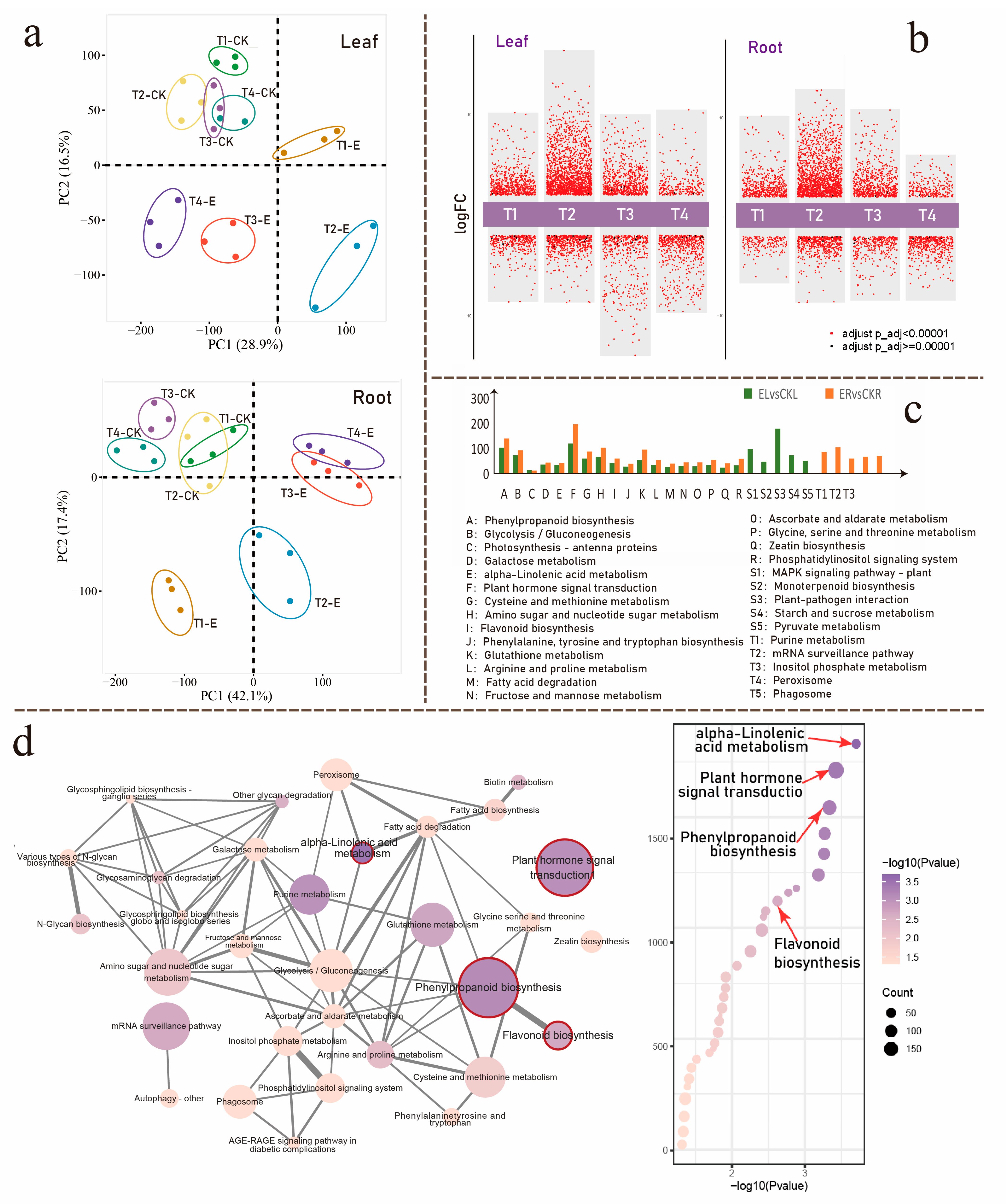
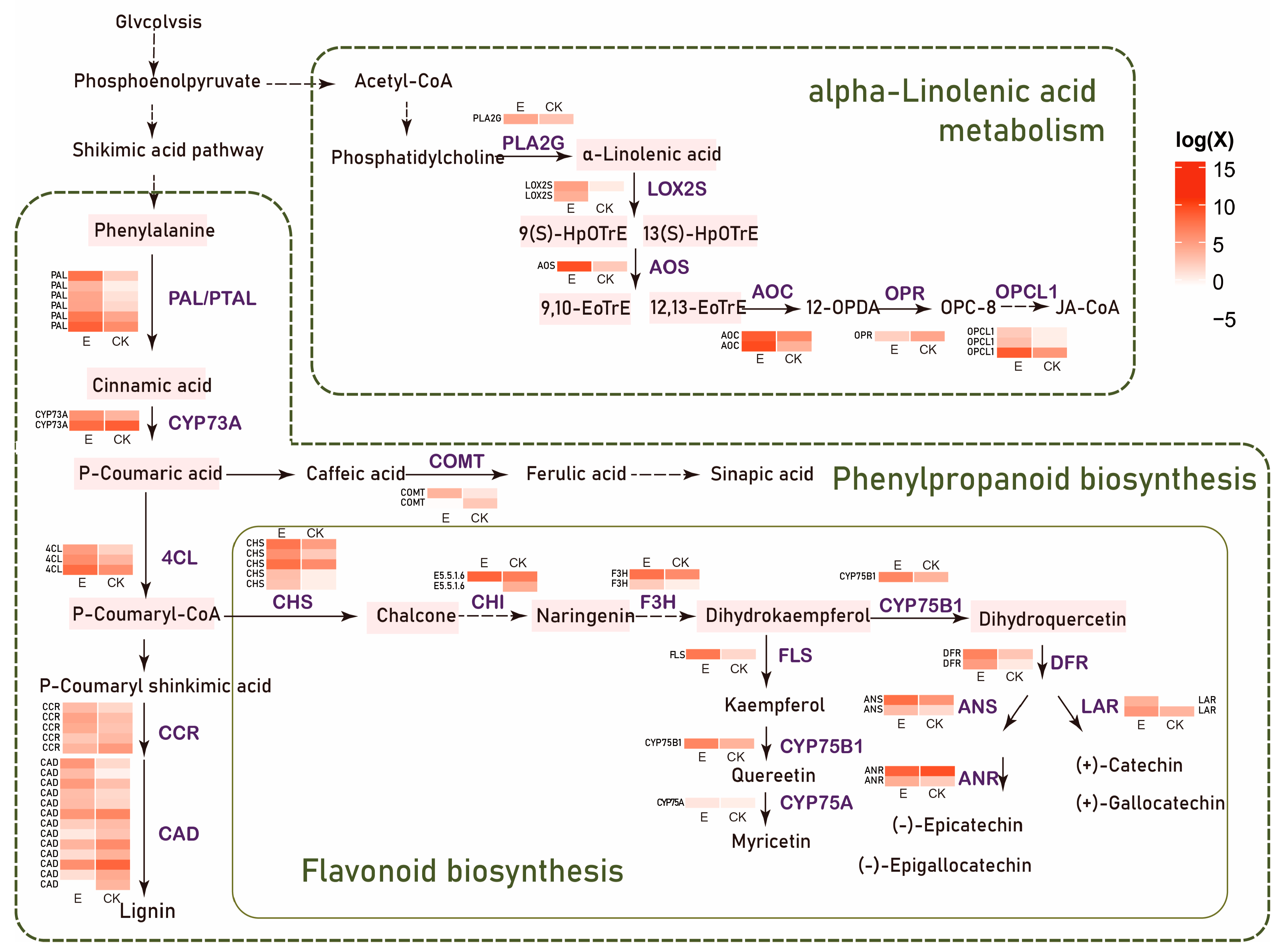
3.2. Ectropis grisescens Infestation Triggers Transcriptomic Reprogramming in Tea Plants
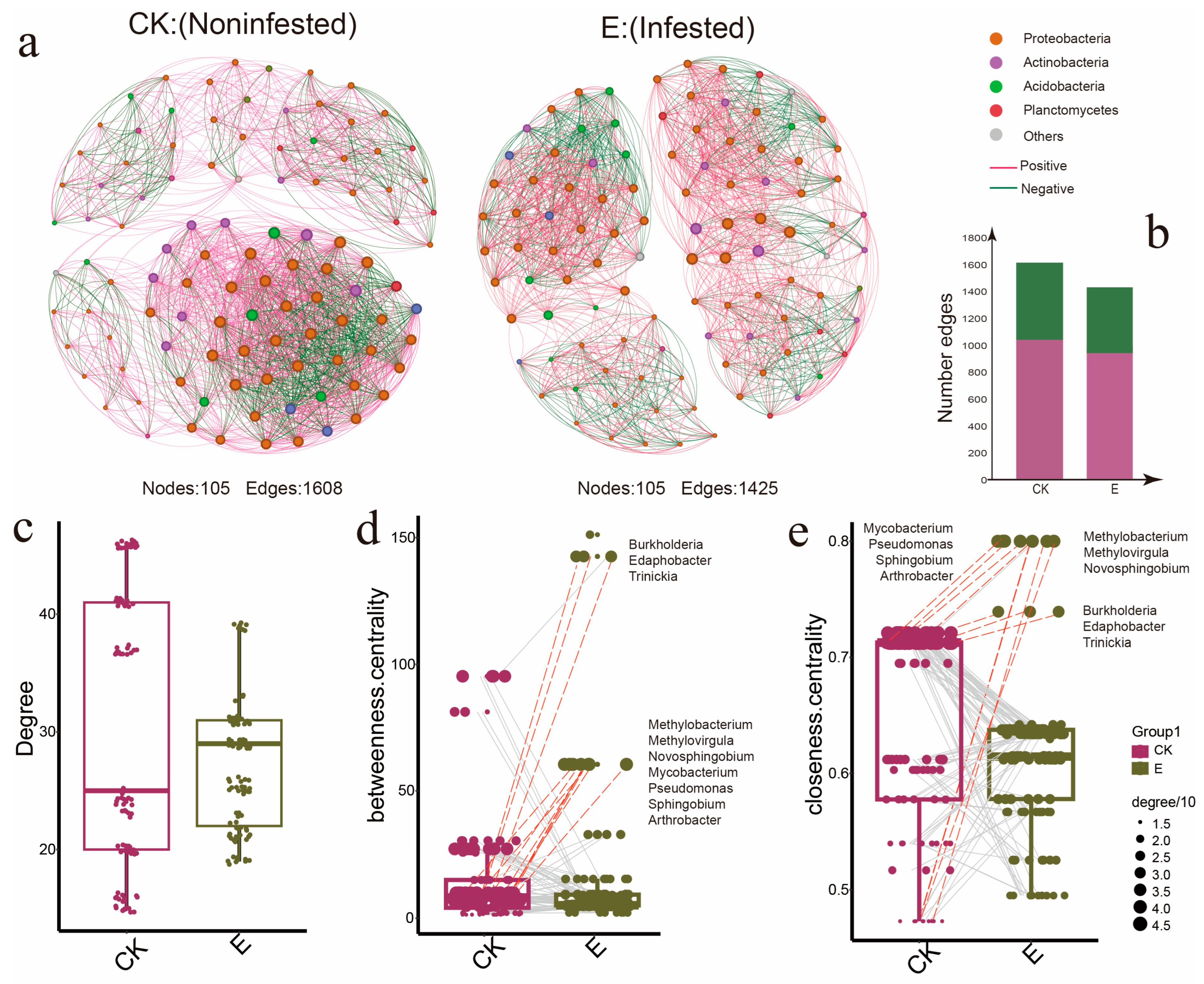
3.3. Ectropis grisescens Infestation Destabilizes Rhizosphere Microbiota Co-Occurrence Networks
3.4. Ectropis grisescens Infestation Affects Tea Rhizosphere Microbiome Function
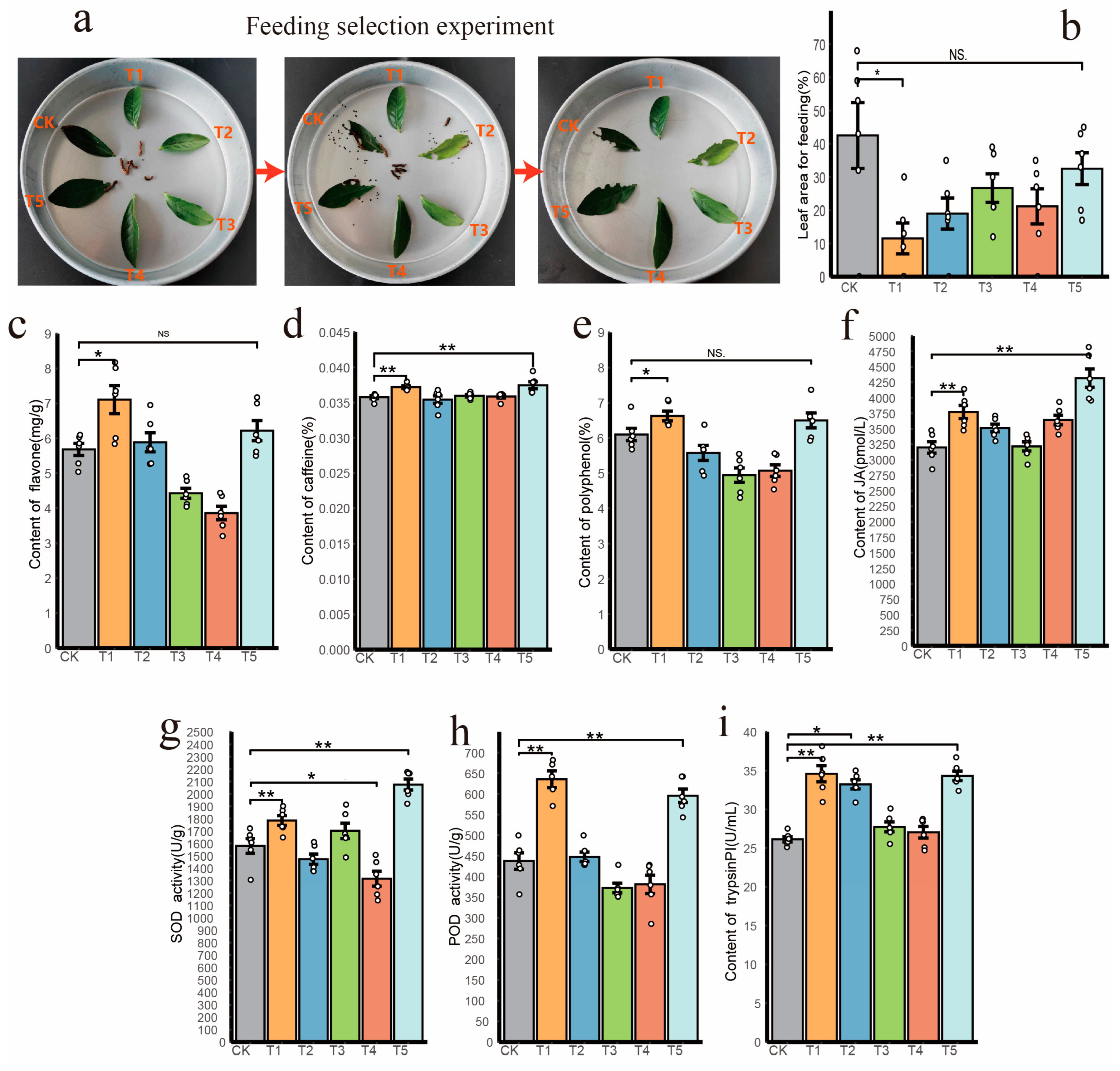
3.5. Burkholderia Positively Enhance Plant Pest-Resistant Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jing, T.; Qian, X.; Du, W.; Gao, T.; Li, D.; Guo, D.; He, F.; Yu, G.; Li, S.; Schwab, W. Herbivore-induced volatiles influence moth preference by increasing the β-ocimene emission of neighbouring tea plants. Plant Cell Environ. 2021, 44, 3667–3680. [Google Scholar] [CrossRef]
- Heil, M. Herbivore-Induced Plant Volatiles: Targets, Perception and Unanswered Questions; Wiley Online Library: Hoboken, NJ, USA, 2014. [Google Scholar]
- Dicke, M.; Baldwin, I.T. The evolutionary context for herbivore-induced plant volatiles: Beyond the ‘cry for help’. Trends Plant Sci. 2010, 15, 167–175. [Google Scholar] [CrossRef]
- Turlings, T.C.; Erb, M. Tritrophic interactions mediated by herbivore-induced plant volatiles: Mechanisms, ecological relevance, and application potential. Annu. Rev. Entomol. 2018, 63, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, T.; Jiang, Y.; Yang, Y.; Zhang, L.; Jiang, Z.; Wei, C.; Wan, X.; Yang, H. Alteration of local and systemic amino acids metabolism for the inducible defense in tea plant (Camellia sinensis) in response to leaf herbivory by Ectropis oblique. Arch. Biochem. Biophys. 2020, 683, 108301. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Baldwin, I.T. New insights into plant responses to the attack from insect herbivores. Annu. Rev. Genet. 2010, 44, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Biere, A.; Goverse, A. Plant-mediated systemic interactions between pathogens, parasitic nematodes, and herbivores above-and belowground. Annu. Rev. Phytopathol. 2016, 54, 499–527. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, Q.; Chen, Q.; He, Y.; He, W.; Geng, J.; Zhang, Y.; Zhou, Y.; Zhu, Z.-R. Plants attacked above-ground by leaf-mining flies change below-ground microbiota to enhance plant defense. Hortic. Res. 2024, 11, uhae121. [Google Scholar] [CrossRef]
- García-Cristobal, J.; García-Villaraco, A.; Ramos, B.; Gutierrez-Mañero, J.; Lucas, J. Priming of pathogenesis related-proteins and enzymes related to oxidative stress by plant growth promoting rhizobacteria on rice plants upon abiotic and biotic stress challenge. J. Plant Physiol. 2015, 188, 72–79. [Google Scholar] [CrossRef]
- Hashem, A.; Tabassum, B.; Abd_Allah, E.F. Bacillus subtilis: A plant-growth promoting rhizobacterium that also impacts biotic stress. Saudi J. Biol. Sci. 2019, 26, 1291–1297. [Google Scholar] [CrossRef]
- Lee, B.; Lee, S.; Ryu, C.-M. Foliar aphid feeding recruits rhizosphere bacteria and primes plant immunity against pathogenic and non-pathogenic bacteria in pepper. Ann. Bot. 2012, 110, 281–290. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.-C.; Marchesi, J.R.; Mougel, C.; Selosse, M.-A. Host-microbiota interactions: From holobiont theory to analysis. Microbiome 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Sthultz, C.M.; Whitham, T.G.; Kennedy, K.; Deckert, R.; Gehring, C.A. Genetically based susceptibility to herbivory influences the ectomycorrhizal fungal communities of a foundation tree species. New Phytol. 2009, 184, 657–667. [Google Scholar] [CrossRef]
- Kula, A.A.; Hartnett, D.C.; Wilson, G.W. Effects of mycorrhizal symbiosis on tallgrass prairie plant–herbivore interactions. Ecol. Lett. 2005, 8, 61–69. [Google Scholar] [CrossRef]
- Pineda, A.; Zheng, S.-J.; van Loon, J.J.; Pieterse, C.M.; Dicke, M. Helping plants to deal with insects: The role of beneficial soil-borne microbes. Trends Plant Sci. 2010, 15, 507–514. [Google Scholar] [CrossRef]
- Sun, X. The Effects of Holotrichia Parallela Larva Damage on Rhizosphere Bacterial Community of Peanut and Preliminary Exploration of Its Mechanism; Chinese Academy of Agricultural Sciences: Beijing, China, 2019. [Google Scholar]
- Kim, B.; Song, G.C.; Ryu, C.-M. Root exudation by aphid leaf infestation recruits root-associated Paenibacillus spp. to lead plant insect susceptibility. J. Microbiol. Biotechnol. 2016, 26, 549–557. [Google Scholar] [CrossRef]
- Kong, H.G.; Kim, B.K.; Song, G.C.; Lee, S.; Ryu, C.-M. Aboveground whitefly infestation-mediated reshaping of the root microbiota. Front. Microbiol. 2016, 7, 1314. [Google Scholar] [CrossRef]
- French, E.; Kaplan, I.; Enders, L. Foliar aphid herbivory alters the tomato rhizosphere microbiome, but initial soil community determines the legacy effects. Front. Sustain. Food Syst. 2021, 5, 629684. [Google Scholar] [CrossRef]
- Singh, B.K.; Trivedi, P.; Egidi, E.; Macdonald, C.A.; Delgado-Baquerizo, M. Crop microbiome and sustainable agriculture. Nat. Rev. Microbiol. 2020, 18, 601–602. [Google Scholar] [CrossRef]
- Xiong, Y.; Shao, S.; Li, D.; Liu, H.; Xie, W.; Huang, W.; Li, J.; Nie, C.; Zhang, J.; Hong, Y. Exploring the Impact of Tea (Camellia sinensis (L.) O. Ktze.)/Trachelospermum jasminoides (Lindl.) Lem. Intercropping on Soil Health and Microbial Communities. Agronomy 2024, 14, 1261. [Google Scholar] [CrossRef]
- Lu, H.; Yang, Y.-Q.; Yu, J.; Li, Q.-K.; Huang, Y.-Z.; Li, Y.-C.; Chen, Q.-Z.; Wan, X.-C.; Guo, F. (Z, Z, Z)-3, 6, 9-nonadecadiene, a potential inhibitor of sex pheromone of Ectropis grisescens Warren (Lepidoptera: Geometridae): Electroantennogram test, wind tunnel, and in silico study. bioRxiv 2022. bioRxiv:2002.514740. [Google Scholar]
- Chen, S.; Zhang, L.; Cai, X.; Bian, L.; Luo, Z.; Li, Z.; Ge, L.; Chen, Z.; Xin, Z. (Z)-3-Hexenol induces tea defense against Ectropis grisescens in the field. Russ. J. Plant Physiol. 2020, 67, 572–580. [Google Scholar] [CrossRef]
- Siegwart, M.; Graillot, B.; Blachere Lopez, C.; Besse, S.; Bardin, M.; Nicot, P.C.; Lopez-Ferber, M. Resistance to bio-insecticides or how to enhance their sustainability: A review. Front. Plant Sci. 2015, 6, 381. [Google Scholar] [CrossRef] [PubMed]
- Berling, M.; Blachere-Lopez, C.; Soubabere, O.; Lery, X.; Bonhomme, A.; Sauphanor, B.; Lopez-Ferber, M. Cydia pomonella granulovirus genotypes overcome virus resistance in the codling moth and improve virus efficiency by selection against resistant hosts. Appl. Environ. Microbiol. 2009, 75, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, D.; Muthumeena, K.; Lavanya, N.; Suresh, S.; Rajendran, L.; Raguchander, T.; Samiyappan, R. Pseudomonas-induced defence molecules in rice plants against leaffolder (Cnaphalocrocis medinalis) pest. Pest Manag. Sci. Former. Pestic. Sci. 2007, 63, 714–721. [Google Scholar] [CrossRef]
- Ling, S.; Zhao, Y.; Sun, S.; Zheng, D.; Sun, X.; Zeng, R.; Chen, D.; Song, Y. Enhanced anti-herbivore defense of tomato plants against Spodoptera litura by their rhizosphere bacteria. BMC Plant Biol. 2022, 22, 254. [Google Scholar] [CrossRef]
- Pineda, A.; Kaplan, I.; Bezemer, T.M. Steering soil microbiomes to suppress aboveground insect pests. Trends Plant Sci. 2017, 22, 770–778. [Google Scholar] [CrossRef]
- Verma, P.P.; Shelake, R.M.; Das, S.; Sharma, P.; Kim, J.-Y. Plant growth-promoting rhizobacteria (PGPR) and fungi (PGPF): Potential biological control agents of diseases and pests. In Microbial Interventions in Agriculture and Environment: Volume 1: Research Trends, Priorities and Prospects; Springer: Singapore, 2019; pp. 281–311. [Google Scholar]
- Rabari, A.; Ruparelia, J.; Jha, C.K.; Sayyed, R.Z.; Mitra, D.; Priyadarshini, A.; Senapati, A.; Panneerselvam, P.; Mohapatra, P.K.D. Articulating beneficial rhizobacteria-mediated plant defenses through induced systemic resistance: A review. Pedosphere 2023, 33, 556–566. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Tang, L.; Hou, Y.; Wang, P.; Yang, H.; Wei, C.-L. Differential transcriptome analysis of leaves of tea plant (Camellia sinensis) provides comprehensive insights into the defense responses to Ectropis oblique attack using RNA-Seq. Funct. Integr. Genom. 2016, 16, 383–398. [Google Scholar] [CrossRef]
- Tai, Y.; Wei, C.; Yang, H.; Zhang, L.; Chen, Q.; Deng, W.; Wei, S.; Zhang, J.; Fang, C.; Ho, C. Transcriptomic and phytochemical analysis of the biosynthesis of characteristic constituents in tea (Camellia sinensis) compared with oil tea (Camellia oleifera). BMC Plant Biol. 2015, 15, 190. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Shi, L.; Gong, D.; Zhang, S.; Zhao, Q.; Zhan, D.; Vasseur, L.; Wang, Y.; Yu, J. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 2021, 53, 1250–1259. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 36, D13–D21. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Anesini, C.; Ferraro, G.E.; Filip, R. Total polyphenol content and antioxidant capacity of commercially available tea (Camellia sinensis) in Argentina. J. Agric. Food Chem. 2008, 56, 9225–9229. [Google Scholar] [CrossRef]
- Rehman, R.; Ashraf, S. Analysis of caffeine contents in commercial beverages and tea samples of Pakistan using UV/Visible spectrometry. Bulg. Chem. Commun. 2017, 49, 823–828. [Google Scholar]
- Bae, I.K.; Ham, H.M.; Jeong, M.H.; Kim, D.H.; Kim, H.J. Simultaneous determination of 15 phenolic compounds and caffeine in teas and mate using RP-HPLC/UV detection: Method development and optimization of extraction process. Food Chem. 2015, 172, 469–475. [Google Scholar] [CrossRef]
- Kenasa, G.; Assefa, F.; Habteselassie, M.Y. Phenotypic and genotypic characteristics of cowpea rhizobia from soils of Ethiopia. Afr. J. Biotechnol. 2018, 17, 1299–1312. [Google Scholar]
- Boss, B.L.; Wanees, A.E.; Zaslow, S.J.; Normile, T.G.; Izquierdo, J.A. Comparative genomics of the plant-growth promoting bacterium Sphingobium sp. strain AEW4 isolated from the rhizosphere of the beachgrass Ammophila breviligulata. BMC Genom. 2022, 23, 508. [Google Scholar] [CrossRef]
- Aguirre, M.; Kiegle, E.; Leo, G.; Ezquer, I. Carbohydrate reserves and seed development: An overview. Plant Reprod. 2018, 31, 263–290. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Gong, J.; Luo, S.; Zuo, Y.; Shen, Y. Role of gamma-aminobutyric acid in plant defense response. Metabolites 2023, 13, 741. [Google Scholar] [CrossRef]
- Sharma, S.S.; Dietz, K.-J. The significance of amino acids and amino acid-derived molecules in plant responses and adaptation to heavy metal stress. J. Exp. Bot. 2006, 57, 711–726. [Google Scholar] [CrossRef]
- Zayed, O.; Hewedy, O.A.; Abdelmoteleb, A.; Ali, M.; Youssef, M.S.; Roumia, A.F.; Seymour, D.; Yuan, Z.-C. Nitrogen journey in plants: From uptake to metabolism, stress response, and microbe interaction. Biomolecules 2023, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lv, J.; Xie, J.; Gan, Y.; Coulter, J.A.; Yu, J.; Li, J.; Wang, J.; Zhang, X. Nitrogen source affects the composition of metabolites in pepper (Capsicum annuum L.) and regulates the synthesis of capsaicinoids through the GOGAT–GS pathway. Foods 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Friman, J.; Karssemeijer, P.N.; Haller, J.; de Kreek, K.; van Loon, J.J.; Dicke, M. Shoot and root insect herbivory change the plant rhizosphere microbiome and affects cabbage–insect interactions through plant–soil feedback. New Phytol. 2021, 232, 2475–2490. [Google Scholar] [CrossRef]
- Ye, G.-Y.; Xiao, Q.; Chen, M.; Chen, X.-x.; Yuan, Z.-j.; Stanley, D.W.; Hu, C. Tea: Biological control of insect and mite pests in China. Biol. Control 2014, 68, 73–91. [Google Scholar] [CrossRef]
- Wang, F.; Wei, Y.; Yan, T.; Wang, C.; Chao, Y.; Jia, M.; An, L.; Sheng, H. Sphingomonas sp. Hbc-6 alters physiological metabolism and recruits beneficial rhizosphere bacteria to improve plant growth and drought tolerance. Front. Plant Sci. 2022, 13, 1002772. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, S.K.; Singh, M.K.; Singh, V.K.; Modi, A.; Singh, P.K.; Kumar, A. Plant growth–promoting rhizobacteria and their functional role in salinity stress management. In Abatement of Environmental Pollutants; Elsevier: Amsterdam, The Netherlands, 2020; pp. 151–160. [Google Scholar]
- Dargiri, S.A.; Naeimi, S. Plant growth-promoting Rhizobacteria in management of biotic and abiotic stresses. In Updates on Rhizobacteria; IntechOpen: London, UK, 2024. [Google Scholar]
- Kerchev, P.I.; Fenton, B.; Foyer, C.H.; Hancock, R.D. Plant responses to insect herbivory: Interactions between photosynthesis, reactive oxygen species and hormonal signalling pathways. Plant Cell Environ. 2012, 35, 441–453. [Google Scholar] [CrossRef]
- Lin, J.; Dai, H.; Yuan, J.; Tang, C.; Ma, B.; Xu, J. Arsenic-induced enhancement of diazotrophic recruitment and nitrogen fixation in Pteris vittata rhizosphere. Nat. Commun. 2024, 15, 10003. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, U.A.; Joseph, C.M.; Phillips, D.A. Flavonoids released naturally from alfalfa seeds enhance growth rate of Rhizobium meliloti. Plant Physiol. 1991, 95, 797–803. [Google Scholar] [CrossRef]
- Deng, L.; Luo, L.; Li, Y.; Wang, L.; Zhang, J.; Zi, B.; Ye, C.; Liu, Y.; Huang, H.; Mei, X. Autotoxic ginsenoside stress induces changes in root exudates to recruit the beneficial Burkholderia strain B36 as revealed by transcriptomic and metabolomic approaches. J. Agric. Food Chem. 2023, 71, 4536–4549. [Google Scholar] [CrossRef]
- Li, Y.; Feng, F.; Mu, Q.e.; Li, M.; Ma, L.; Wan, Q.; Jousset, A.; Liu, C.; Yu, X. Foliar Spraying of Chlorpyrifos Triggers Plant Production of Linolenic Acid Recruiting Rhizosphere Bacterial Sphingomonas sp. Environ. Sci. Technol. 2023, 57, 17312–17323. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Song, Z.; Li, Z.; Qiao, R.; Zhang, P.; Ding, C.; Xie, J.; Chen, Y.; Guo, H. Populus root exudates are associated with rhizosphere microbial communities and symbiotic patterns. Front. Microbiol. 2022, 13, 1042944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, J.; Khashi u Rahman, M.; Wu, F. The impact of root exudates, volatile organic compounds, and common mycorrhizal networks on root system architecture in root-root interactions. J. Plant Interact. 2022, 17, 685–694. [Google Scholar] [CrossRef]
- Van Der Heijden, M.G.; Hartmann, M. Networking in the plant microbiome. PLoS Biol. 2016, 14, e1002378. [Google Scholar] [CrossRef]
- Hernandez, D.J.; David, A.S.; Menges, E.S.; Searcy, C.A.; Afkhami, M.E. Environmental stress destabilizes microbial networks. ISME J. 2021, 15, 1722–1734. [Google Scholar] [CrossRef]
- Estrada-De Los Santos, P.; Bustillos-Cristales, R.O.; Caballero-Mellado, J. Burkholderia, a genus rich in plant-associated nitrogen fixers with wide environmental and geographic distribution. Appl. Environ. Microbiol. 2001, 67, 2790–2798. [Google Scholar] [CrossRef]
- Caballero-Mellado, J.; Onofre-Lemus, J.; Estrada-De Los Santos, P.; Martínez-Aguilar, L. The tomato rhizosphere, an environment rich in nitrogen-fixing Burkholderia species with capabilities of interest for agriculture and bioremediation. Appl. Environ. Microbiol. 2007, 73, 5308–5319. [Google Scholar] [CrossRef]
- Garau, G.; Yates, R.J.; Deiana, P.; Howieson, J.G. Novel strains of nodulating Burkholderia have a role in nitrogen fixation with papilionoid herbaceous legumes adapted to acid, infertile soils. Soil Biol. Biochem. 2009, 41, 125–134. [Google Scholar] [CrossRef]
- Elliott, G.N.; Chen, W.-M.; Chou, J.-H.; Wang, H.-C.; Sheu, S.-Y.; Perin, L.; Reis, V.M.; Moulin, L.; Simon, M.F.; Bontemps, C. Burkholderia phymatum is a highly effective nitrogen-fixing symbiont of Mimosa spp. and fixes nitrogen ex planta. New Phytol. 2007, 173, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Madhaiyan, M.; Saravanan, V.S.; Wirth, J.S.; Alex, T.H.H.; Kim, S.-J.; Weon, H.-Y.; Kwon, S.-W.; Whitman, W.B.; Ji, L. Sphingomonas palmae sp. nov. and Sphingomonas gellani sp. nov., endophytically associated phyllosphere bacteria isolated from economically important crop plants. Antonie Van Leeuwenhoek 2020, 113, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- Madhaiyan, M.; Alex, T.H.H.; Ngoh, S.T.; Prithiviraj, B.; Ji, L. Leaf-residing Methylobacterium species fix nitrogen and promote biomass and seed production in Jatropha curcas. Biotechnol. Biofuels 2015, 8, 222. [Google Scholar] [CrossRef]
- Wolińska, A.; Kuźniar, A.; Zielenkiewicz, U.; Banach, A.; Izak, D.; Stępniewska, Z.; Błaszczyk, M. Metagenomic analysis of some potential nitrogen-fixing bacteria in arable soils at different formation processes. Microb. Ecol. 2017, 73, 162–176. [Google Scholar] [CrossRef]
- Arrobas, M.; Correia, C.M.; Rodrigues, M.Â. Methylobacterium symbioticum Applied as a Foliar Inoculant Was Little Effective in Enhancing Nitrogen Fixation and Lettuce Dry Matter Yield. Sustainability 2024, 16, 4512. [Google Scholar] [CrossRef]
- Fang, W.; Yan, D.; Wang, Q.; Huang, B.; Ren, Z.; Wang, X.; Wang, X.; Li, Y.; Ouyang, C.; Migheli, Q. Changes in the abundance and community composition of different nitrogen cycling groups in response to fumigation with 1, 3-dichloropropene. Sci. Total Environ. 2019, 650, 44–55. [Google Scholar] [CrossRef]
- Pang, Z.; Mao, X.; Zhou, S.; Yu, S.; Liu, G.; Lu, C.; Wan, J.; Hu, L.; Xu, P. Microbiota-mediated nitrogen fixation and microhabitat homeostasis in aerial root-mucilage. Microbiome 2023, 11, 85. [Google Scholar] [CrossRef]
- Zimmer, S.; Messmer, M.; Haase, T.; Piepho, H.-P.; Mindermann, A.; Schulz, H.; Habekuß, A.; Ordon, F.; Wilbois, K.-P.; Heß, J. Effects of soybean variety and Bradyrhizobium strains on yield, protein content and biological nitrogen fixation under cool growing conditions in Germany. Eur. J. Agron. 2016, 72, 38–46. [Google Scholar] [CrossRef]
- Hungria, M.; Menna, P.; Delamuta, J.R.M. Bradyrhizobium, the ancestor of all rhizobia: Phylogeny of housekeeping and nitrogen-fixation genes. In Biological Nitrogen Fixation; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 191–202. [Google Scholar]
- Huang, X.; Zhou, W.; Zhang, Y.; Yang, Q.; Yang, B.; Liang, T.; Ling, J.; Dong, J. Keystone PGPR ecological effect: An inoculation case study of diazotrophic Novosphingobium sp. N034 on mangrove plant Kandelia obovate. Appl. Soil Ecol. 2024, 202, 105567. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.N.; Qi, Y.; Goren, E.; Chiniquy, D.; Sheflin, A.M.; Tringe, S.G.; Prenni, J.E.; Liu, P.; Schachtman, D.P. Root-associated bacterial communities and root metabolite composition are linked to nitrogen use efficiency in sorghum. mSystems 2024, 9, e01190-23. [Google Scholar] [CrossRef] [PubMed]
- Mattson, W.J. Herbivory in relation to plant nitrogen content. Annu. Rev. Ecol. Syst. 1980, 11, 119–161. [Google Scholar] [CrossRef]
- Nalam, V.J.; Shah, J.; Nachappa, P. Emerging role of roots in plant responses to aboveground insect herbivory. Insect Sci. 2013, 20, 286–296. [Google Scholar] [CrossRef]
- Ankala, A.; Luthe, D.; Williams, W.; Wilkinson, J. Integration of ethylene and jasmonic acid signaling pathways in the expression of maize defense protein Mir1-CP. Mol. Plant-Microbe Interact. 2009, 22, 1555–1564. [Google Scholar] [CrossRef]
- Mithöfer, A.; Boland, W. Plant defense against herbivores: Chemical aspects. Annu. Rev. Plant Biol. 2012, 63, 431–450. [Google Scholar] [CrossRef]
- Liao, Y.; Tan, H.; Jian, G.; Zhou, X.; Huo, L.; Jia, Y.; Zeng, L.; Yang, Z. Herbivore-induced (Z)-3-hexen-1-ol is an airborne signal that promotes direct and indirect defenses in tea (Camellia sinensis) under light. J. Agric. Food Chem. 2021, 69, 12608–12620. [Google Scholar] [CrossRef]
- Graham, J.; McNicol, R.; Greig, K. Towards genetic based insect resistance in strawberry using the cowpea trypsin inhibitor gene. Ann. Appl. Biol. 1995, 127, 163–173. [Google Scholar] [CrossRef]
- Boulter, D.; Gatehouse, A.; Hilder, V. Use of cowpea trypsin inhibitor (CpTI) to protect plants against insect predation. Biotechnol. Adv. 1989, 7, 489–497. [Google Scholar] [CrossRef]
- Ramaroson, M.-L.; Koutouan, C.; Helesbeux, J.-J.; Le Clerc, V.; Hamama, L.; Geoffriau, E.; Briard, M. Role of phenylpropanoids and flavonoids in plant resistance to pests and diseases. Molecules 2022, 27, 8371. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Navarro, J.A.; Vega, F.E.; Karaoz, U.; Hao, Z.; Jenkins, S.; Lim, H.C.; Kosina, P.; Infante, F.; Northen, T.R.; Brodie, E.L. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 2015, 6, 7618. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kaur, I.; Kariyat, R. The multifunctional roles of polyphenols in plant-herbivore interactions. Int. J. Mol. Sci. 2021, 22, 1442. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Chen, W.; Fang, X.; Li, D.; Xiong, Y.; Xie, W.; Chen, Q.; You, Y.; Lin, C.; Wang, Z.; et al. Impact of Ectropis grisescens Warren (Lepidoptera: Geometridae) Infestation on the Tea Plant Rhizosphere Microbiome and Its Potential for Enhanced Biocontrol and Plant Health Management. Insects 2025, 16, 412. https://doi.org/10.3390/insects16040412
Liu H, Chen W, Fang X, Li D, Xiong Y, Xie W, Chen Q, You Y, Lin C, Wang Z, et al. Impact of Ectropis grisescens Warren (Lepidoptera: Geometridae) Infestation on the Tea Plant Rhizosphere Microbiome and Its Potential for Enhanced Biocontrol and Plant Health Management. Insects. 2025; 16(4):412. https://doi.org/10.3390/insects16040412
Chicago/Turabian StyleLiu, He, Wei Chen, Xiaohong Fang, Dongliang Li, Yulin Xiong, Wei Xie, Qiulian Chen, Yingying You, Chenchen Lin, Zhong Wang, and et al. 2025. "Impact of Ectropis grisescens Warren (Lepidoptera: Geometridae) Infestation on the Tea Plant Rhizosphere Microbiome and Its Potential for Enhanced Biocontrol and Plant Health Management" Insects 16, no. 4: 412. https://doi.org/10.3390/insects16040412
APA StyleLiu, H., Chen, W., Fang, X., Li, D., Xiong, Y., Xie, W., Chen, Q., You, Y., Lin, C., Wang, Z., Wang, J., Chen, D., Li, Y., Cai, P., Nie, C., & Hong, Y. (2025). Impact of Ectropis grisescens Warren (Lepidoptera: Geometridae) Infestation on the Tea Plant Rhizosphere Microbiome and Its Potential for Enhanced Biocontrol and Plant Health Management. Insects, 16(4), 412. https://doi.org/10.3390/insects16040412