
Complete Mitochondrial Genome of Apis cerana (Hymenoptera: Apidae) from Two Geographical Regions: Insights into Structure and Genetic Differentiation

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
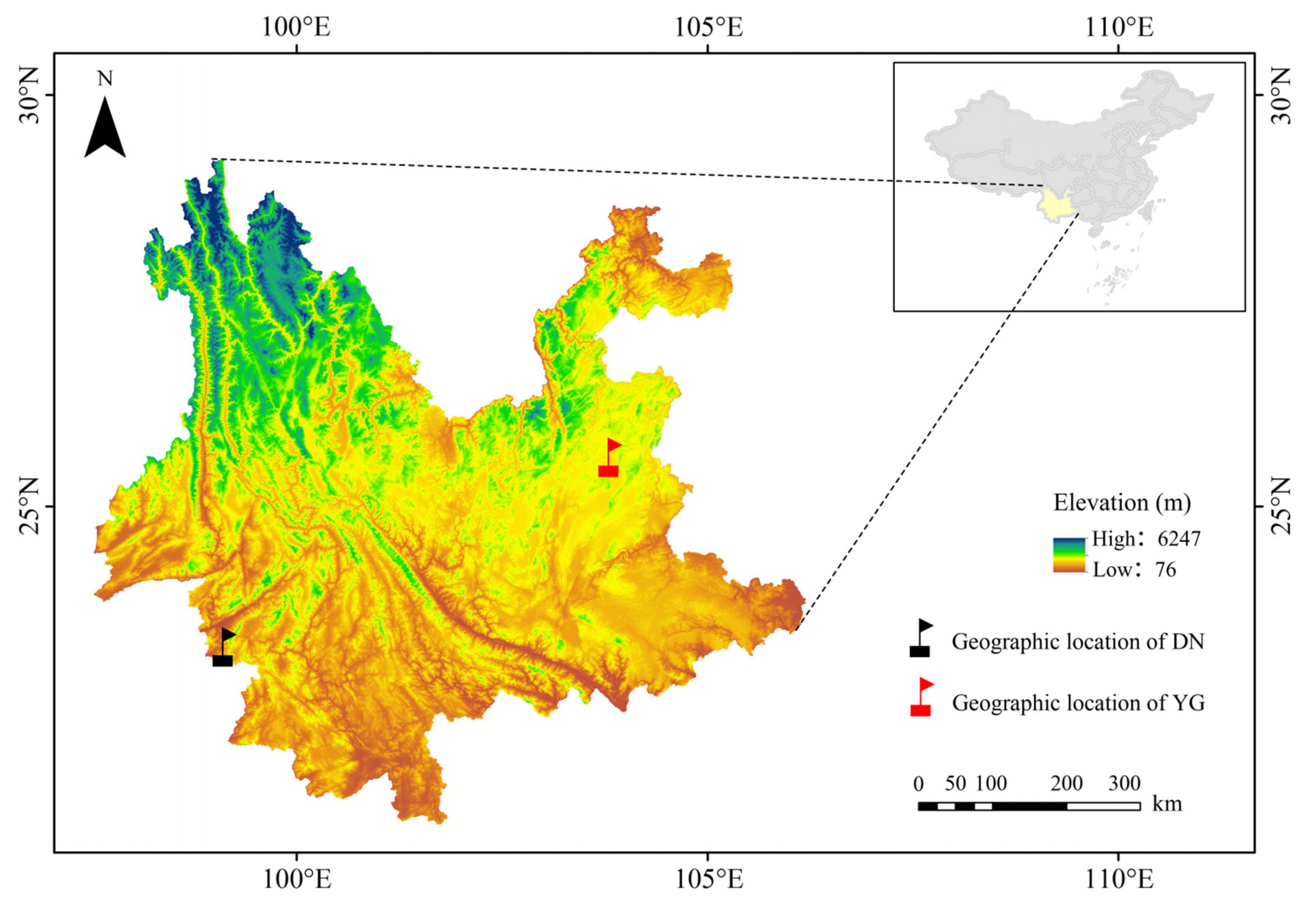
2.1. Sample Collection and DNA Sequencing
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Mitochondrial Genome Sequence Analysis
2.4. Phylogenetic Analyses
3. Results
3.1. Mitogenome Organization and Base Composition
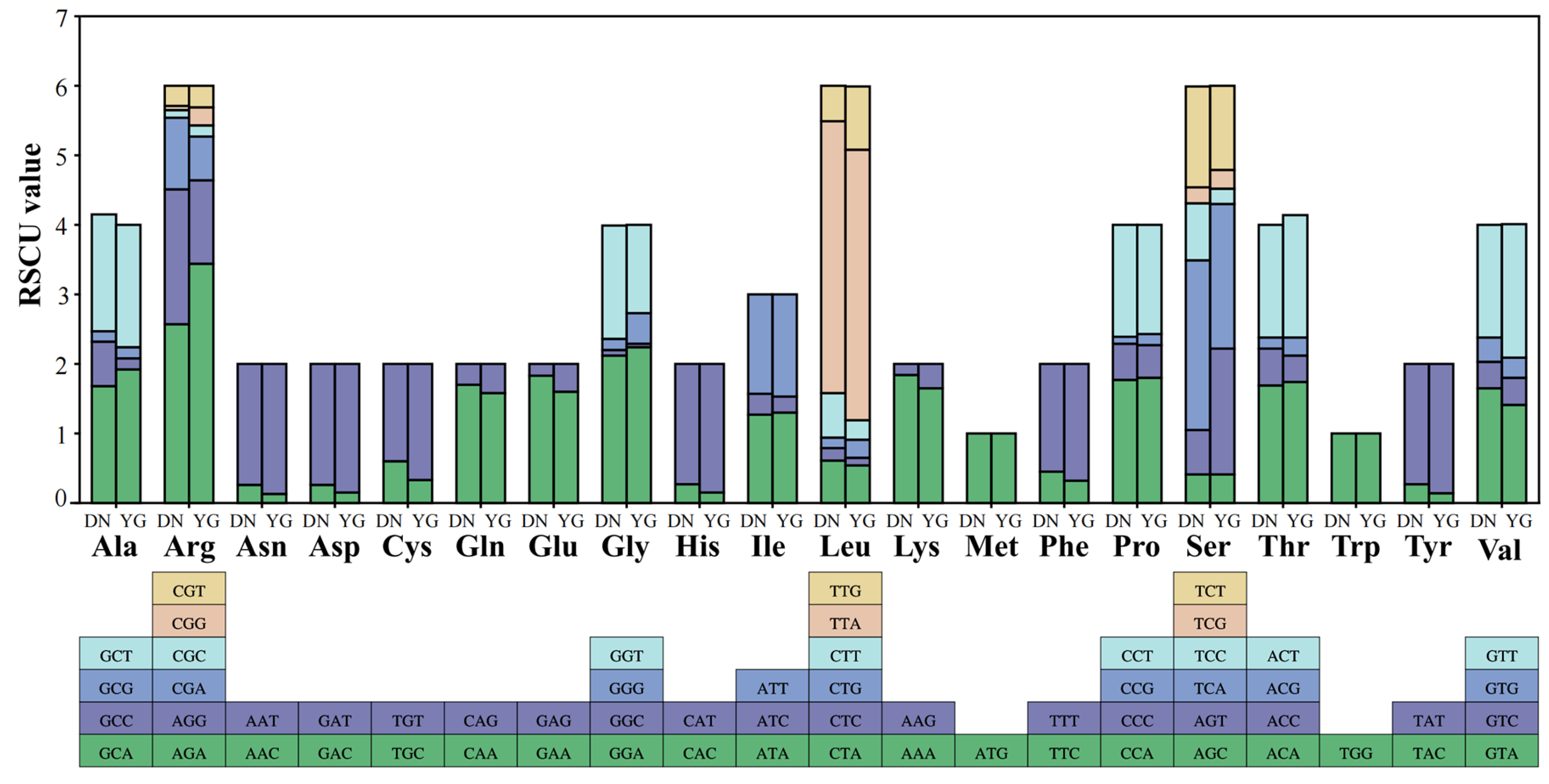
3.2. Protein-Coding Genes
3.3. Transfer RNAs and Ribosomal RNAs
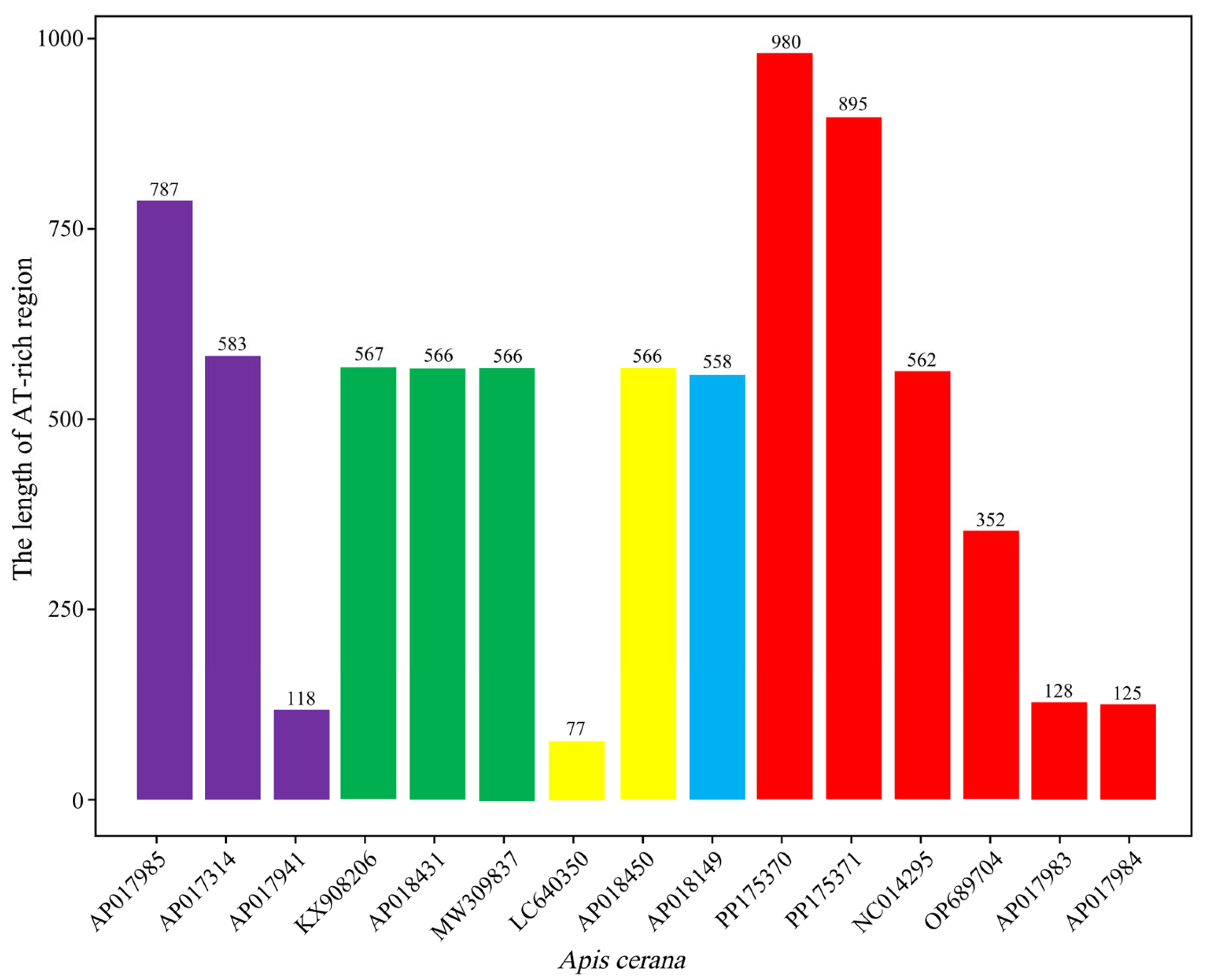
3.4. AT-Rich Region
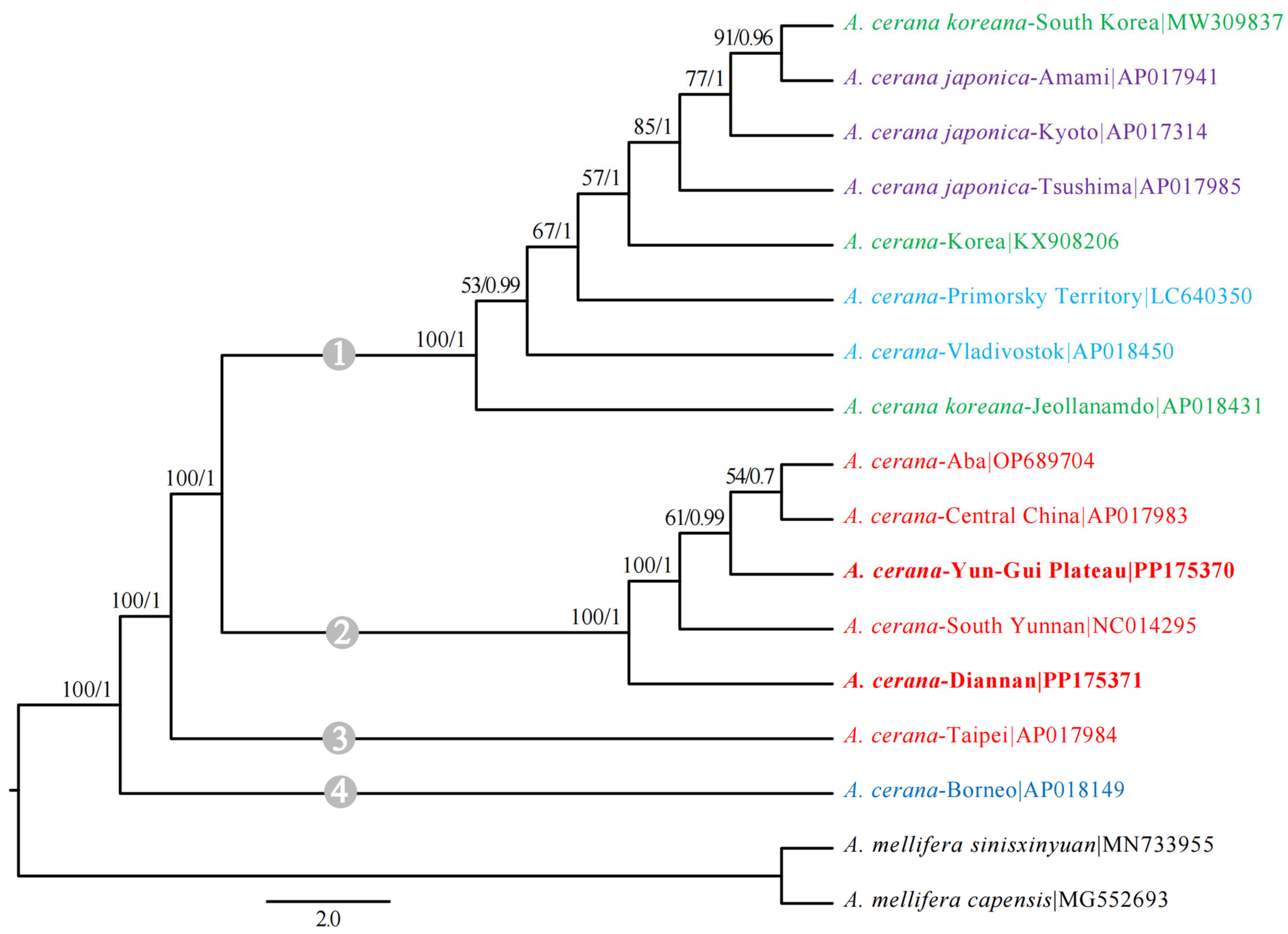
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lan, L.; Shi, P.; Song, H.L.; Tang, X.Y.; Zhou, J.Y.; Yang, J.D.; Yang, M.X.; Xu, J.S. De Novo genome assembly of Chinese plateau honeybee unravels intraspecies genetic diversity in the eastern honeybee, Apis cerana. Insects 2021, 12, 891. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.C.; Shi, W.; Tang, J.; Gao, J.L.; Liu, F.; Shan, J.Q.; Chen, X.; Chen, C. Morphometrical analyses revealed high diversity of the eastern honey bee (Apis cerana) in mountains and islands in China. J. Apic. Res. 2023, 62, 647–655. [Google Scholar] [CrossRef]
- Li, G.L.; Zhao, H.; Guo, D.Z.; Liu, Z.G.; Wang, H.F.; Sun, Q.H.; Liu, Q.X.; Xu, B.H.; Guo, X.Q. Distinct molecular impact patterns of abamectin on Apis mellifera ligustica and Apis cerana cerana. Ecotoxicol. Environ. Saf. 2022, 232, 113242. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.M.S.; Leventon, J.; Rau, A.; Borgemeister, C.; von Wehrden, H. A review of ecosystem service benefits from wild bees across social contexts. Ambio 2017, 46, 456–467. [Google Scholar] [CrossRef]
- Li, Y.C.; Chao, T.L.; Fan, Y.H.; Lou, D.L.; Wang, G.Z. Population genomics and morphological features underlying the adaptive evolution of the eastern honey bee (Apis cerana). BMC Genom. 2019, 20, 869. [Google Scholar]
- Qiu, L.F.; Dong, J.X.; Li, X.G.; Parey, S.H.; Tan, K.; Orr, M.; Majeed, A.; Zhang, X.; Luo, S.Q.; Zhou, X.G.; et al. Defining honeybee subspecies in an evolutionary context warrants strategized conservation. Zool. Res. 2023, 44, 483–493. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, W.F.; Wang, H.F.; Xv, B.H.; Liu, Z.G. Research progress on molecular markers in honeybee. J. Bee 2022, 42, 33–37. [Google Scholar]
- Xu, L.Q.; Yang, J.D.; Lai, K. The Complete Mitochondrial Genome of the Cavity-Nesting Honeybee, Apis cerana abansis (Insecta: Hymenoptera: Apidae). Cytol. Genet. 2024, 58, 136–141. [Google Scholar] [CrossRef]
- Dowling, D.K.; Wolff, J.N.; Cooley, L. Evolutionary genetics of the mitochondrial genome: Insights from Drosophila. Genetics 2023, 224, iyad036. [Google Scholar] [CrossRef]
- Tang, X.Y.; Yao, Y.X.; Li, Y.H.; Song, H.L.; Luo, R.; Shi, P.; Zhou, Z.Y.; Xu, J.S. Comparison of the mitochondrial genomes of three geographical strains of Apis laboriosa indicates high genetic diversity in the black giant honeybee (Hymenoptera: Apidae). Ecol. Evol. 2023, 13, e9782. [Google Scholar] [CrossRef]
- Yu, Y.L.; Zhou, S.J.; Zhu, X.J.; Xu, X.J.; Wang, W.F.; Zha, L.; Wang, P.; Wang, J.W.; Lai, K.; Wang, S.H.; et al. Genetic differentiation of eastern honey bee (Apis cerana) populations across Qinghai-Tibet plateau-valley landforms. Front. Genet. 2019, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Z.G.; Pan, Q.; Chen, X.; Wang, H.H.; Guo, H.K.; Liu, S.D.; Lu, H.F.; Tian, S.L.; Li, R.Q.; et al. Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n. ssp. Mol. Biol. Evol. 2016, 33, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, H.H.; Liu, Z.G.; Chen, X.; Tang, J.; Meng, F.M.; Shi, W. Population genomics provide insights into the evolution and adaptation of the eastern honey bee (Apis cerana). Mol. Biol. Evol. 2018, 35, 2260–2271. [Google Scholar] [CrossRef]
- Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Ellis, J.D. Mitochondrial genome diversity and population structure of two western honey bee subspecies in the Republic of South Africa. Sci. Rep. 2018, 8, 1333. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Park, J.; Takahashi, J.; Kwon, H.W. Phylogenetic Uniqueness of Honeybee Apis cerana from the Korean Peninsula Inferred from The Mitochondrial, Nuclear, and Morphological Data. J. Apic. Sci. 2018, 62, 189–214. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Youn, H.G.; Lee, M.; Kim, K.W.; Proshchalykin, M.Y.; Lelej, A.S.; Takahashi, J.; Kwon, H.W. Phylogenetic relationships of Russian Far-East Apis cerana with other North Asian populations. J. Apic. Sci. 2019, 63, 289–314. [Google Scholar] [CrossRef]
- Fan, X.Y.; Tang, D.; Liao, Y.H.; Li, P.D.; Zhang, Y.; Wang, M.X.; Liang, F.; Wang, X.; Gao, Y.; Wen, L.; et al. Single-cell RNA-seq analysis of mouse preimplantation embryos by third-generation sequencing. PLoS Biol. 2020, 18, e3001017. [Google Scholar] [CrossRef]
- He, X.J.; Barron, A.B.; Yang, L.; Chen, H.; He, Y.Z.; Zhang, L.Z.; Huang, Q.; Wang, Z.L.; Wu, X.B.; Yan, W.Y.; et al. Extent and complexity of RNA processing in honey bee queen and worker caste development. iScience 2022, 25, 104301. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Concepcion, G.T.; Feng, X.W.; Zhang, H.W.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Jarvis, E.D.; Fedrigo, O.; Koepfli, K.P.; Urban, L.; Gemmell, N.J.; Li, H. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 2022, 40, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Xiang, C.Y.; Gao, F.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, 2–87. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic. Acids. Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Wakamiya, T.; Fujiwara, A.; Washitani, I.; Takahashi, J.I. Complete mitochondrial genome of the honeybee Apis cerana native to two remote islands in Japan. Conserv. Genet. Resour. 2017, 9, 557–560. [Google Scholar] [CrossRef]
- Takahashi, J.; Wakamiya, T.; Kiyoshi, T.; Uchiyama, H.; Yajima, S.; Kimura, K.; Nomura, T. The complete mitochondrial genome of the Japanese honeybee, Apis cerana japonica (Insecta: Hymenoptera: Apidae). Mitochondrial DNA Part B Resour. 2016, 1, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Tingek, S.; Takahashi, J. The complete mitochondrial genome of the cavity-nesting honeybee, Apis cerana (Insecta: Hymenoptera: Apidae) from Borneo. Mitochondrial DNA Part B Resour. 2017, 2, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Liu, G.H.; Dong, X.; Lin, R.Q.; Song, H.Q.; Huang, S.Y.; Yuan, Z.G.; Zhao, G.H.; Zhu, X.Q. The complete mitochondrial genome of the Asiatic cavity-nesting honeybee Apis cerana (Hymenoptera: Apidae). PLoS ONE 2011, 6, e23008. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.S.; Zhao, C.X.; Chen, F.; Liu, Y.H.; Zhang, S.Z.; Wu, H.; Zhang, L.S.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef]
- Wang, X.Y.; Li, D.F.; Li, H.; Wang, J.J.; Li, Y.J.; Dai, R.H. Comparison of mitogenomes of three Petalocephala species (Hemiptera: Cicadellidae: Ledrinae) and their phylogenetic analysis. Arch. Insect Biochem. Physiol. 2022, 111, e21902. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Liu, X.Y.; Yang, D. The first mitochondrial genome for caddisfly (insecta: Trichoptera) with phylogenetic implications. Int. J. Biol. Sci. 2013, 10, 53–63. [Google Scholar] [CrossRef]
- Chen, Z.T.; Du, Y.Z. Rearrangement of mitochondrial genome in insects. J. Environ. Entomol. 2016, 38, 843–851. [Google Scholar]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genom. 2010, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.N.; Cen, Y.J.; Wallis, C.M.; Trumble, J.T.; Prager, S.; Yokomi, R.; Zheng, Z.; Deng, X.L.; Chen, J.C.; Liang, G.W.; et al. The complete mitochondrial genome sequence of Bactericera cockerelli and comparison with three other Psylloidea species. PLoS ONE 2016, 11, e0155318. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.F.; Tang, L.; Chen, J.; Zhang, X.Y.; Easy, R.H.; You, P. The mitogenome of freshwater loach Homatula laxiclathra (Teleostei: Nemacheilidae) with phylogenetic analysis of Nemacheilidae. Ecol. Evol. 2020, 10, 5990–6000. [Google Scholar] [CrossRef]
- Zhang, C.H.; Wang, Y.L.; Chen, H.W.; Huang, J. Comparative mitochondrial genomes between the genera amiota and phortica (Diptera: Drosophilidae) with evolutionary insights into D-Loop sequence variability. Genes 2023, 14, 1240. [Google Scholar] [CrossRef]
- Fredens, J.; Wang, K.; de la Torre, D.; Funke, L.F.H.; Robertson, W.E.; Christova, Y.; Chia, T.; Schmied, W.H.; Dunkelmann, D.L.; Beránek, V.; et al. Total synthesis of Escherichia coli with a recoded genome. Nature 2019, 569, 514–518. [Google Scholar] [CrossRef]
- Komar, A.A. A code within a code: How codons fine-tune protein folding in the cell. Biochemistry 2021, 86, 976–991. [Google Scholar] [CrossRef]
- Peng, G.X.; Mao, X.L.; Cao, Y.; Yao, S.Y.; Li, Q.R.; Chen, X.; Wang, E.D.; Zhou, X.L. RNA granule-clustered mitochondrial aminoacyl-tRNA synthetases form multiple complexes with the potential to fine-tune tRNA aminoacylation. Nucleic Acids Res. 2022, 50, 12951–12968. [Google Scholar] [CrossRef]
- Andargie, M.; Congyi, Z. Genome-wide analysis of codon usage in sesame (Sesamum indicum L.). Heliyon 2022, 8, e08687. [Google Scholar] [CrossRef]
- Huang, Y.X.; Wang, S.; Gao, Y.Q.; Chen, J.H.; Wang, X.L.; Li, R.J. Comparison of mitochondrial genome and development of specific PCR primers for identifying two scuticociliates, Pseudocohnilembus persalinus and Uronema marinum. Parasites Vectors 2021, 14, 318. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.G.; Bao, Z.Y.; Mou, C.X.; Chen, Z.H.; Zhao, J.W. Comprehensive analysis of codon usage on porcine astrovirus. Viruses 2020, 12, 991. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y. Comparative analysis of resistance of forest resources to rain and snow freezing disasters in Yunnan Province. J. Green Sci. Technol. 2019, 223–226, 236. [Google Scholar]
- Hu, Q.Q.; Pan, Y.Q.; Xia, H.L.; Yu, K.X.; Yao, Y.; Guan, F. Species identification of caviar based on multiple DNA barcoding. Molecules 2023, 28, 5046. [Google Scholar] [CrossRef]
- Suryawanshi, V.; Talke, I.N.; Weber, M.; Eils, R.; Brors, B.; Clemens, S.; Krämer, U. Between-species differences in gene copy number are enriched among functions critical for adaptive evolution in Arabidopsis halleri. BMC Genom. 2016, 17, 1034. [Google Scholar] [CrossRef]
- Liu, J.; Bu, C.P.; Wipfler, B.; Liang, A.P. Comparative analysis of the mitochondrial genomes of Callitettixini Spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the AT-rich region but reveals the presence of short conservative elements at the tribal level. PLoS ONE 2014, 9, e109140. [Google Scholar] [CrossRef]
- Rajewska, M.; Kowalczyk, L.; Konopa, G.; Konieczny, I. Specific mutations within the AT-rich region of a plasmid replication origin affect either origin opening or helicase loading. Proc. Natl. Acad. Sci. USA 2008, 105, 11134–11139. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Luo, W.T.; Song, W.F.; Liang, C.; Zhang, X.W. Morphology of Apis cerana in the Yunnan-Guizhou plateau and bees in south of Yunnan. Apic. China 2014, 65, 12–15. [Google Scholar]
- Kitnya, N.; Prabhudev, M.V.; Bhatta, C.P.; Pham, T.H.; Nidup, T.; Megu, K.; Chakravorty, J.; Brockmann, A.; Otis, G.W. Geographical distribution of the giant honey bee Apis laboriosa Smith, 1871 (Hymenoptera, Apidae). Zookeys 2020, 951, 67–81. [Google Scholar] [CrossRef]
- Tang, X.Y.; Song, H.L.; Shi, P.; Zhang, X.Y.; Tang, Z.H.; Wang, W.F.; Zha, L.; Chen, X.L.; Zhou, Z.Y.; Xv, J.S. Whole-genome resequencing reveals the genetic diversity and adaptive evolution of Apis cerana (Hymenoptera: Apidae) on the eastern and southeastern edges of the Qinghai-Tibet Plateau. Acta Entomol. Sin. 2022, 65, 638–647. [Google Scholar]
- Liu, N.N.; Liu, H.M.; Ju, Y.; Li, X.G.; Li, Y.; Wang, T.J.; He, J.M.; Niu, Q.S.; Xing, X.M. Geometric morphology and population genomics provide insights into the adaptive evolution of Apis cerana in Changbai Mountain. BMC Genom. 2022, 23, 64. [Google Scholar]
- Fang, F.; Chen, X.S.; Lv, J.; Shi, X.Y.; Feng, X.J.; Wang, Z.; Li, X. Population structure and genetic diversity of Chinese Honeybee (Apis cerana cerana) in central China. Genes 2022, 13, 1007. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Naeem, M.; Ali, H.; Shakeel, M.; Kuang, H.O.; Zhang, Z.; Sun, C. Genome sequence of the Asian honeybee in Pakistan sheds light on its phylogenetic relationship with other honeybees. Insects 2021, 12, 652. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Species | GenBank Accession Number | Sampling Locality | Reference |
|---|---|---|---|---|
| 1 | Apis cerana japonica-Tsushima | AP017985.1 | Japan | [34] |
| 2 | Apis cerana japonica-Kyoto | AP017314.1 | Japan | [35] |
| 3 | Apis cerana japonica-Amami | AP017941.1 | Japan | [34] |
| 4 | Apis cerana-Primorsky Territory | LC640350.1 | Russia | Directly Submitted |
| 5 | Apis cerana-Korea | KX908206.1 | Korea | Directly Submitted |
| 6 | Apis cerana koreana-Jeollanamdo | AP018431.1 | Korea | Directly Submitted |
| 7 | Apis cerana koreana-South Korea | MW309837.1 | Korea | Directly Submitted |
| 8 | Apis cerana-Vladivostok | AP018450.1 | Russia | [17] |
| 9 | Apis cerana-Borneo | AP018149.1 | Borneo | [36] |
| 10 | Apis cerana-South Yunnan | NC014295.1 | China | [37] |
| 11 | Apis cerana-Aba | OP689704.1 | China | Directly Submitted |
| 12 | Apis cerana-Central China | AP017983.2 | China | [34] |
| 13 | Apis cerana-Taipei | AP017984.2 | China | [34] |
| 14 | Apis mellifera sinisxinyuan * | MN733955.1 | China | Directly Submitted |
| 15 | Apis mellifera capensis * | MG552693.1 | South Africa | [15] |
| Gene | Start Position | End Position | Length/bp | Intergenic Nucleotides/bp | Initiation Codons | Termination Codons | Direction |
|---|---|---|---|---|---|---|---|
| trnS1(Ser) | 1/1 | 60/60 | 60/60 | J/J | |||
| trnE(Glu) | 64/64 | 129/129 | 66/66 | 3/3 | J/J | ||
| trnM(Met) | 164/164 | 229/229 | 66/66 | 34/34 | J/J | ||
| trnQ(Gln) | 461/461 | 522/522 | 62/62 | 230/231 | J/J | ||
| trnA(Ala) | 523/523 | 588/588 | 66/66 | 0/0 | J/J | ||
| trnI(Ile) | 607/607 | 672/672 | 66/66 | 18/18 | J/J | ||
| ND2 | 673/673 | 1668/1668 | 996/996 | 0/0 | ATT/ATT | TAA/TAA | J/J |
| trnC(Cys) | 1668/1668 | 1733/1733 | 66/66 | −1/−1 | N/N | ||
| trnY(Tyr) | 1739/1739 | 1807/1807 | 69/69 | 5/5 | N/N | ||
| trnW(Trp) | 1824/1824 | 1892/1892 | 69/69 | 16/16 | J/J | ||
| COX1 | 1893/1893 | 3453/3453 | 1561/1561 | 0/0 | ATT/ATT | T(AA)/T(AA) | J/J |
| trnL2(Leu) | 3454/3454 | 3523/3523 | 70/70 | 0/0 | J/J | ||
| COX2 | 3613/3613 | 4291/4291 | 679/679 | 89/89 | ATT/ATT | T(AA)/T(AA) | J/J |
| trnD(Asp) | 4292/4292 | 4359/4359 | 68/68 | 0/0 | J/J | ||
| trnK(Lys) | 4366/4366 | 4437/4437 | 72/72 | 6/6 | J/J | ||
| ATP8 | 4444/4444 | 4605/4605 | 162/162 | 6/6 | ATC/ATC | TAA/TAA | J/J |
| ATP6 | 4587/4587 | 5264/5264 | 678/678 | −19/−19 | ATG/ATG | TAA/TAA | J/J |
| COX3 | 5282/5282 | 6061/6061 | 780/780 | 17/17 | ATG/ATG | TAA/TAA | J/J |
| trnG(Gly) | 6125/6132 | 6191/6198 | 67/67 | 63/70 | J/J | ||
| ND3 | 6192/6199 | 6545/6552 | 354/354 | 0/0 | ATT/ATT | TAA/TAA | J/J |
| trnR(Arg) | 6566/6572 | 6631/6637 | 66/66 | 20/19 | N/N | ||
| trnN(Asn) | 6651/6657 | 6718/6724 | 68/68 | 19/19 | J/J | ||
| trnF(Phe) | 6737/6743 | 6807/6813 | 71/71 | 18/18 | N/N | ||
| ND5 | 6814/6820 | 8481/8487 | 1668/1668 | 6/6 | ATT/ATT | TAA/TAA | N/N |
| trnH(His) | 8483/8489 | 8548/8554 | 66/66 | 1/1 | N/N | ||
| ND4 | 8566/8572 | 9894/9900 | 1329/1329 | 17/17 | ATT/ATT | TAA/TAA | N/N |
| ND4L | 9895/9901 | 10,158/10,164 | 264/264 | 0/0 | ATT/ATT | TAA/TAA | N/N |
| trnT(Thr) | 10,182/10,188 | 10,248/10,254 | 67/67 | 23/23 | J/J | ||
| trnP(Pro) | 10,264/10,270 | 10,341/10,347 | 78/78 | 15/15 | N/N | ||
| ND6 | 10,392/10,398 | 10,904/10,910 | 513/513 | 50/50 | ATT/ATT | TAA/TAA | J/J |
| CYTB | 10,917/10,923 | 12,065/12,071 | 1149/1149 | 12/12 | ATG/ATG | TAA/TAA | J/J |
| trnS2(Ser) | 12,089/12,095 | 12,155/12,161 | 67/67 | 23/23 | J/J | ||
| ND1 | 12,168/12,174 | 13,082/13,088 | 915/915 | 12/12 | ATT/ATT | TAA/TAA | N/N |
| trnL1(Leu) | 13,083/13,089 | 13,151/13,157 | 69/69 | 0/0 | N/N | ||
| large subunitr RNA(lrRNA) | 13,152/13,158 | 14,451/14,456 | 1322/1322 | 0/0 | N/N | ||
| trnV(Val) | 14,452/14,457 | 14,546/14,552 | 67/68 | 0/0 | N/N | ||
| small subunit rRNA(srRNA) | 14,547/14,553 | 15,319/15,324 | 773/773 | 0/0 | N/N | ||
| AT-rich region | 15,320/15,325 | 16,214/16,304 | 895/980 | 0/0 | J/J |
| Region | Length (bp) | A% | T% | C% | G% | A + T% | AT-Skew | GC-Skew | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | |
| ND2 | 996 | 996 | 39.26 | 39.36 | 46.89 | 47.19 | 8.63 | 8.23 | 5.22 | 5.22 | 86.15 | 86.55 | −0.09 | −0.09 | −0.25 | −0.22 |
| COX1 | 1561 | 1561 | 34.66 | 34.66 | 41.38 | 41.19 | 12.88 | 13.07 | 11.08 | 11.08 | 76.04 | 75.85 | −0.09 | −0.09 | −0.05 | −0.08 |
| COX2 | 679 | 679 | 37.85 | 38.44 | 40.65 | 40.35 | 12.37 | 12.67 | 9.13 | 8.54 | 78.5 | 78.79 | −0.04 | −0.02 | −0.15 | −0.19 |
| ATP8 | 162 | 162 | 47.53 | 47.53 | 38.89 | 39.51 | 9.26 | 8.64 | 4.32 | 4.32 | 86.42 | 87.04 | 0.1 | 0.09 | −0.36 | −0.33 |
| ATP6 | 678 | 678 | 36.73 | 36.58 | 47.49 | 47.49 | 10.18 | 10.33 | 5.6 | 5.6 | 84.22 | 84.07 | −0.13 | −0.13 | −0.29 | −0.3 |
| COX3 | 780 | 780 | 36.41 | 35.9 | 44.23 | 44.61 | 10.64 | 10.64 | 8.72 | 8.85 | 80.64 | 80.51 | −0.1 | −0.11 | −0.1 | −0.09 |
| ND3 | 354 | 354 | 37.57 | 37.57 | 48.02 | 47.74 | 9.61 | 9.6 | 4.8 | 5.09 | 85.59 | 85.31 | −0.12 | −0.12 | −0.33 | −0.31 |
| ND5 | 1668 | 1668 | 46.94 | 47.06 | 37.95 | 37.89 | 9.53 | 9.47 | 5.58 | 5.58 | 84.89 | 84.95 | 0.11 | 0.11 | −0.26 | −0.26 |
| ND4 | 1329 | 1329 | 49.13 | 49.06 | 35.89 | 35.97 | 9.49 | 9.41 | 5.49 | 5.56 | 85.02 | 85.03 | 0.16 | 0.15 | −0.27 | −0.26 |
| ND4L | 264 | 264 | 52.65 | 52.65 | 34.47 | 34.47 | 9.47 | 9.47 | 3.41 | 3.41 | 87.12 | 87.12 | 0.21 | 0.21 | −0.47 | −0.47 |
| ND6 | 513 | 513 | 43.08 | 43.08 | 43.08 | 43.27 | 8.58 | 8.38 | 5.26 | 5.27 | 86.16 | 86.35 | 0 | 0 | −0.24 | −0.23 |
| CYTB | 1149 | 1149 | 36.73 | 36.73 | 44.21 | 44.21 | 10.53 | 10.53 | 8.53 | 8.53 | 80.94 | 80.94 | −0.09 | −0.09 | −0.1 | −0.1 |
| ND1 | 915 | 915 | 47.98 | 47.98 | 35.52 | 35.52 | 10.93 | 10.93 | 5.57 | 5.57 | 83.5 | 83.5 | 0.15 | 0.15 | −0.32 | −0.32 |
| AT-rich region | 895 | 980 | 47.71 | 47.45 | 49.72 | 50 | 1.56 | 1.33 | 1.01 | 1.22 | 97.43 | 97.45 | −0.02 | −0.03 | −0.21 | −0.04 |
| Region | Length (bp) | A% | T% | C% | G% | A + T% | AT-Skew | GC-Skew | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | DN | YG | |
| lrRNA | 1322 | 1322 | 42.46 | 42.57 | 40.47 | 40.42 | 11.15 | 11.16 | 5.92 | 5.85 | 82.92 | 82.99 | 0.02 | 0.03 | −0.31 | −0.31 |
| srRNA | 773 | 773 | 37.52 | 37.56 | 43.85 | 43.91 | 12.55 | 12.44 | 6.08 | 6.09 | 81.37 | 81.47 | −0.08 | −0.08 | −0.35 | −0.34 |
| rRNA | 2095 | 2095 | 40.62 | 40.70 | 41.73 | 41.72 | 11.67 | 11.64 | 5.98 | 5.94 | 82.35 | 82.42 | −0.01 | −0.01 | −0.32 | −0.32 |
| tRNA | 1486 | 1487 | 42.33 | 42.43 | 44.95 | 44.92 | 7.40 | 7.40 | 5.32 | 5.25 | 87.28 | 87.35 | −0.03 | −0.03 | −0.16 | −0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Su, R.; Zhu, R.; Ding, G.; Guo, Z.; Du, L.; Huang, J. Complete Mitochondrial Genome of Apis cerana (Hymenoptera: Apidae) from Two Geographical Regions: Insights into Structure and Genetic Differentiation. Insects 2024, 15, 960. https://doi.org/10.3390/insects15120960
Chen Y, Su R, Zhu R, Ding G, Guo Z, Du L, Huang J. Complete Mitochondrial Genome of Apis cerana (Hymenoptera: Apidae) from Two Geographical Regions: Insights into Structure and Genetic Differentiation. Insects. 2024; 15(12):960. https://doi.org/10.3390/insects15120960
Chicago/Turabian StyleChen, Yuhui, Runlang Su, Rui Zhu, Guiling Ding, Zhanbao Guo, Lin Du, and Jiaxing Huang. 2024. "Complete Mitochondrial Genome of Apis cerana (Hymenoptera: Apidae) from Two Geographical Regions: Insights into Structure and Genetic Differentiation" Insects 15, no. 12: 960. https://doi.org/10.3390/insects15120960
APA StyleChen, Y., Su, R., Zhu, R., Ding, G., Guo, Z., Du, L., & Huang, J. (2024). Complete Mitochondrial Genome of Apis cerana (Hymenoptera: Apidae) from Two Geographical Regions: Insights into Structure and Genetic Differentiation. Insects, 15(12), 960. https://doi.org/10.3390/insects15120960