
Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Rearing of S. frugiperda
2.2. Experimental Design
2.3. Collection of Tissue Samples and DNA Extraction
2.4. Sequencing of 16S rRNA Gene
2.5. Microbiome Analyses
2.6. Statistical Analysis
3. Results
3.1. Sequencing Data of 16S rRNA
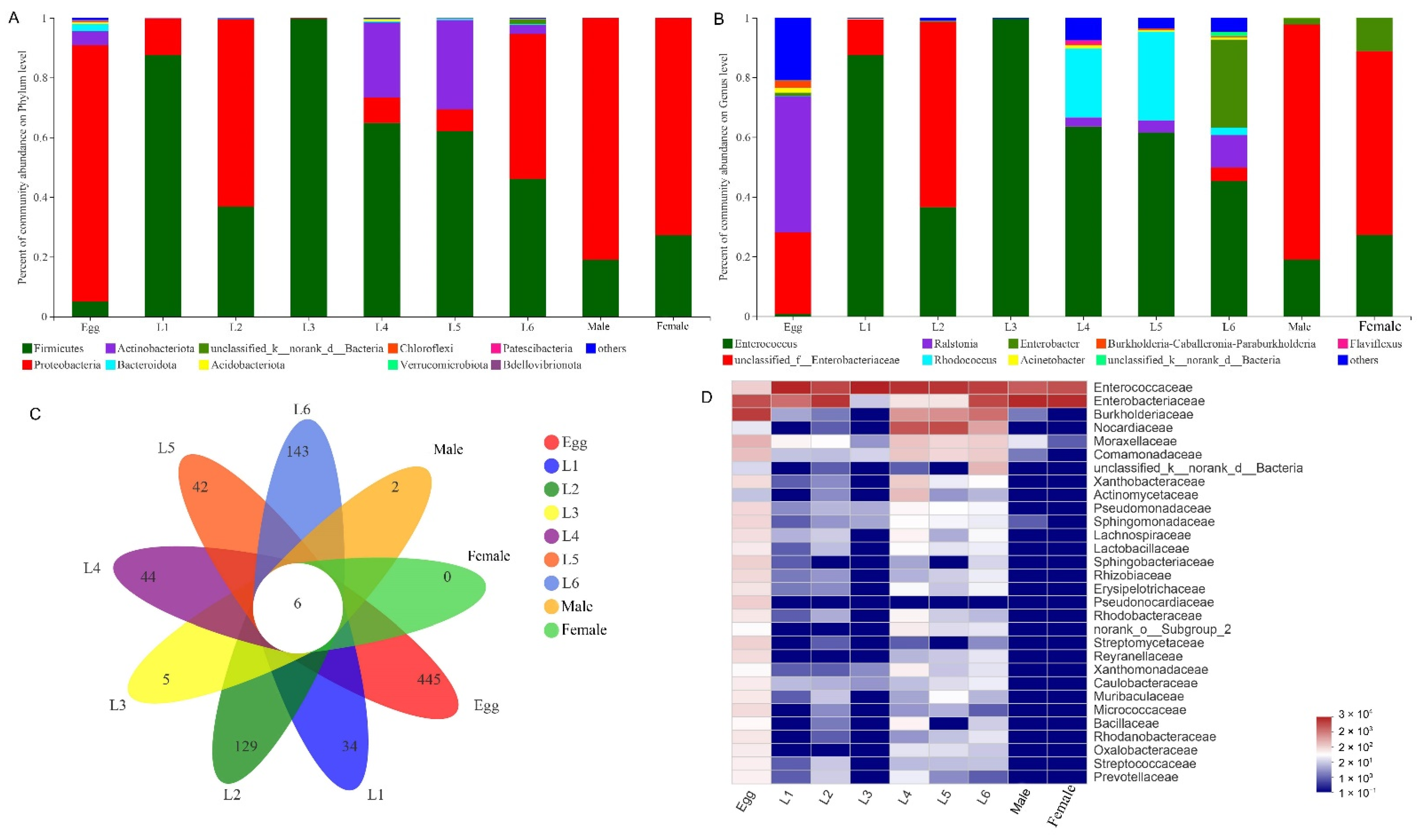
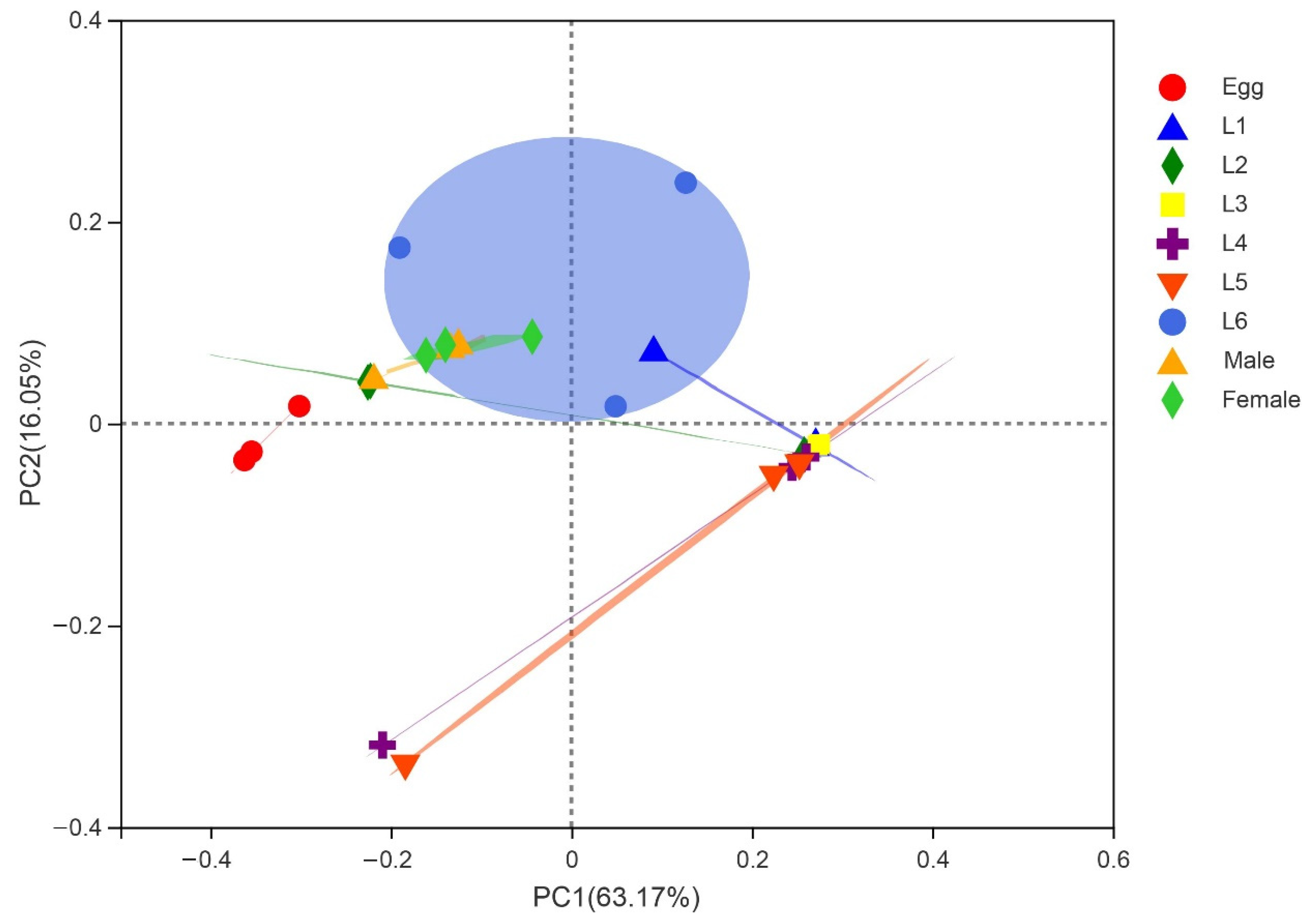
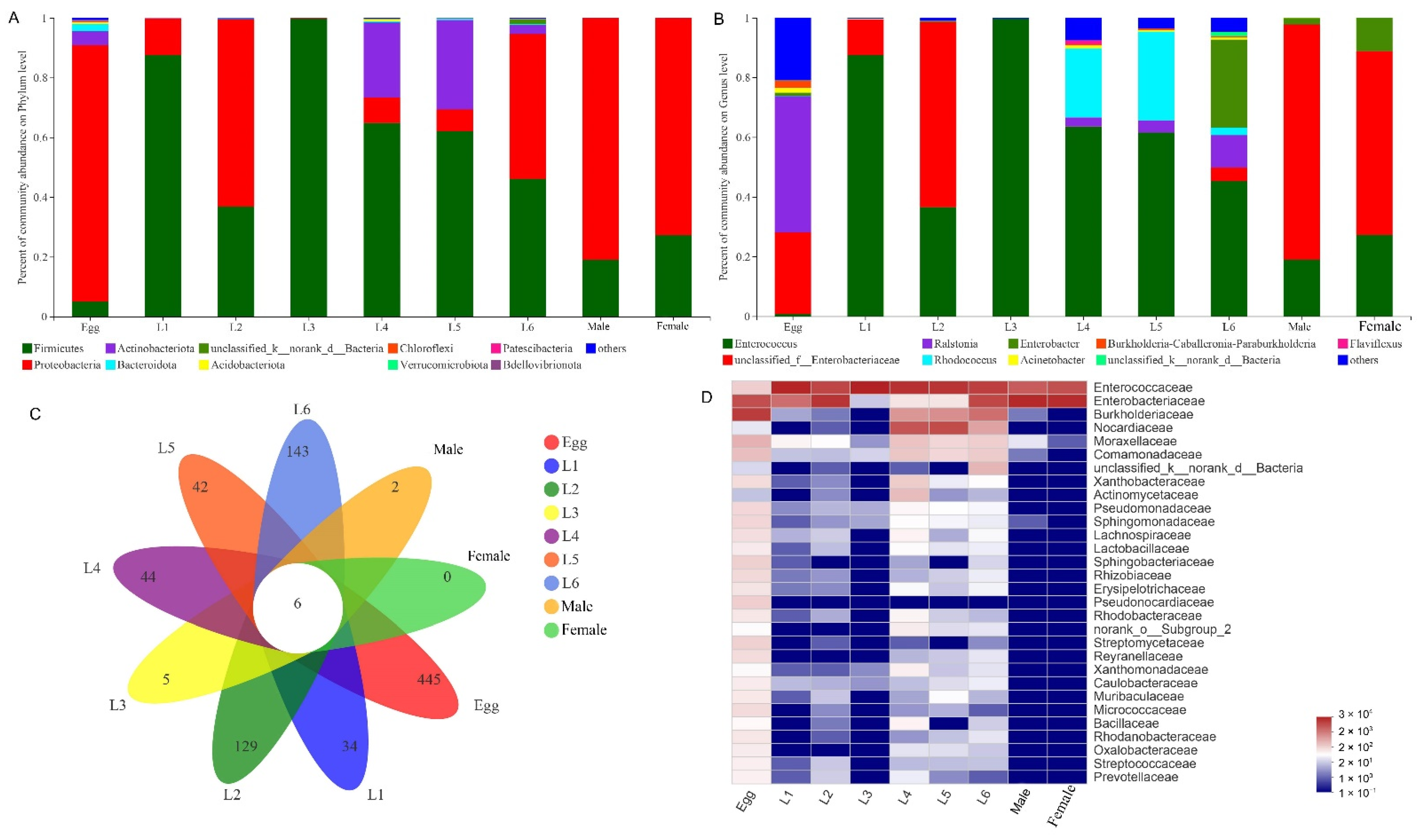
3.2. Gut Microbiota Composition of S. frugiperda across Different Developmental Stages
3.3. Common and Unique Microbes among All Developmental Stages of S. frugiperda
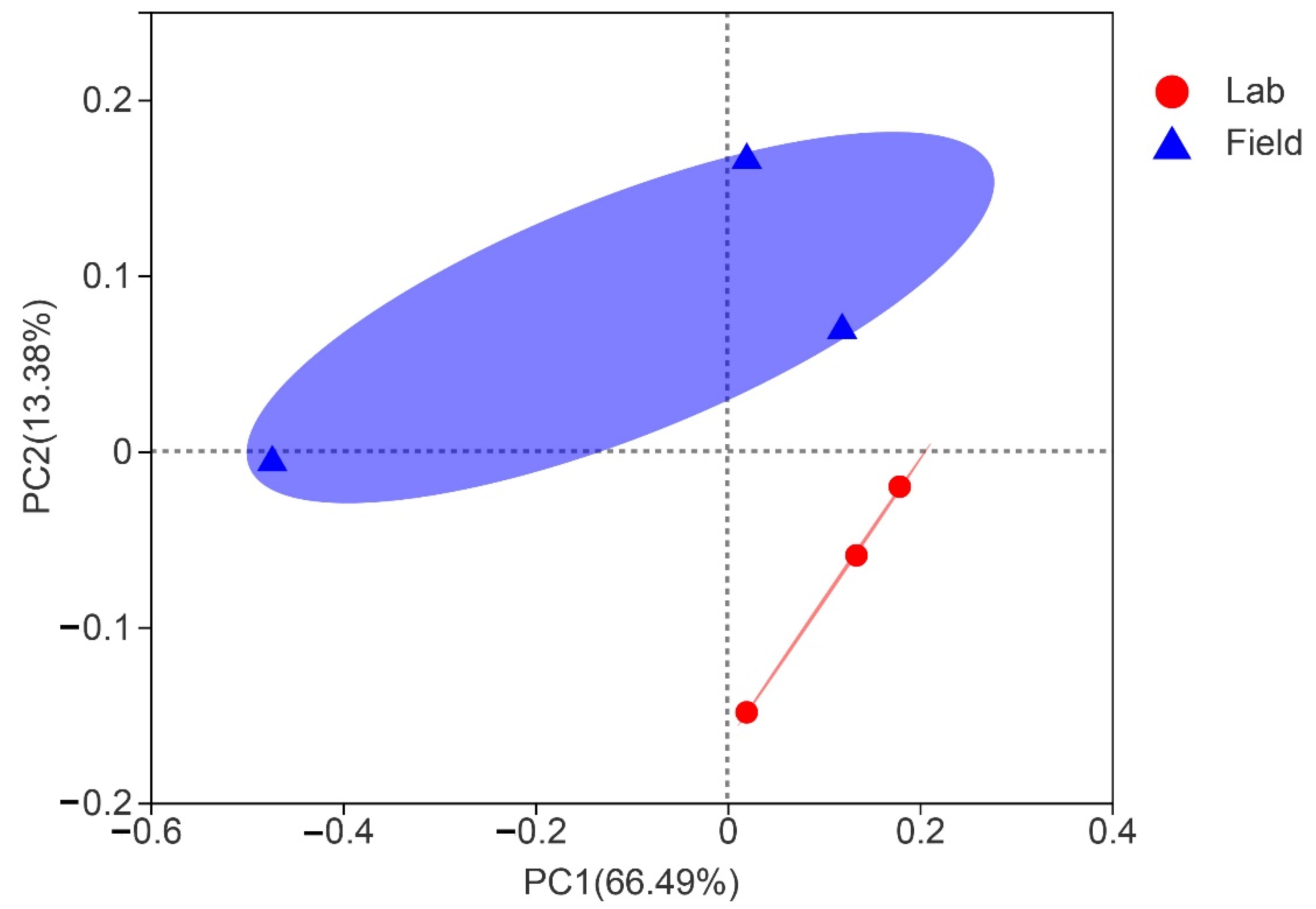
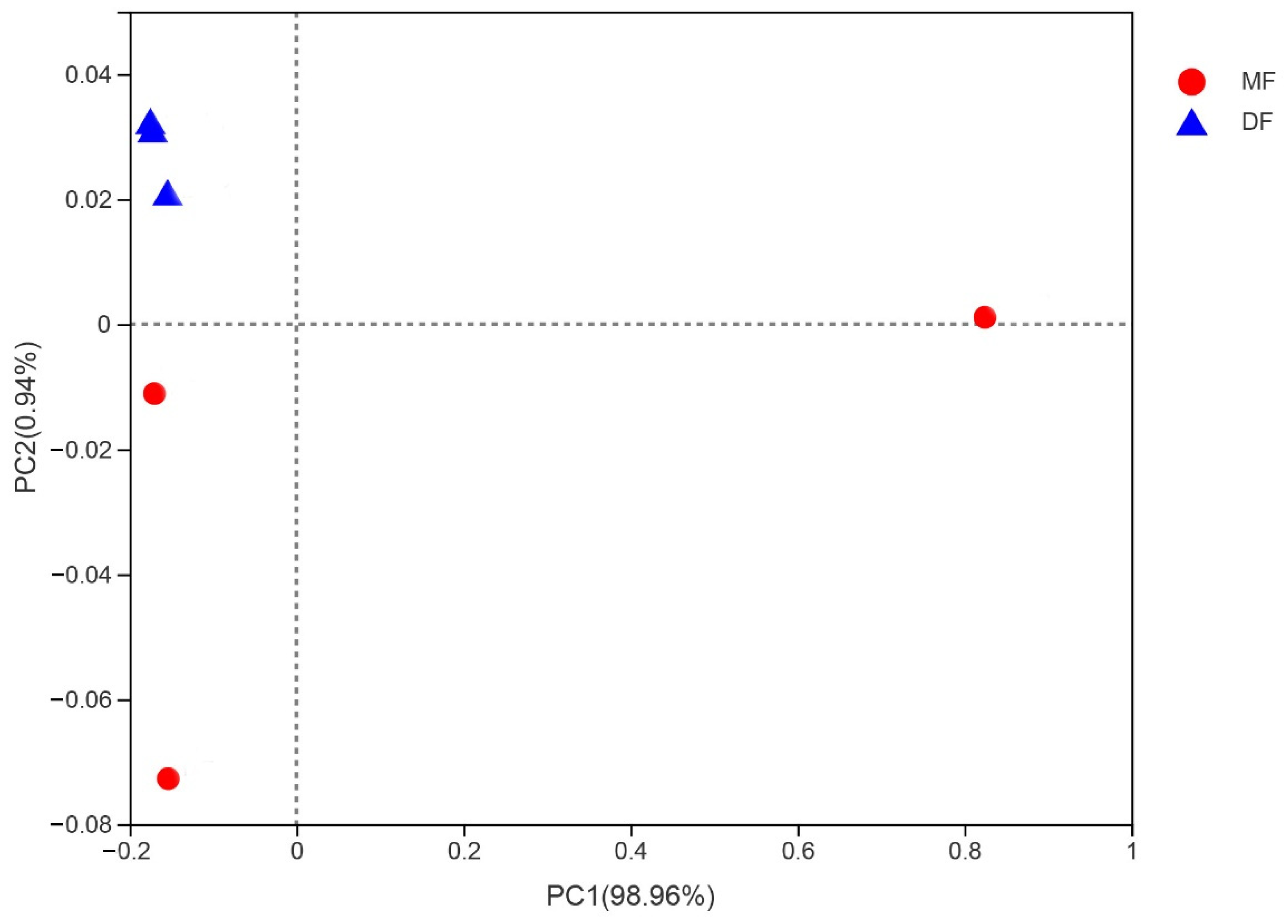
3.4. Comparison of Gut Bacterial Communities of S. frugiperda Associated with Different Environmental Habitats of Host
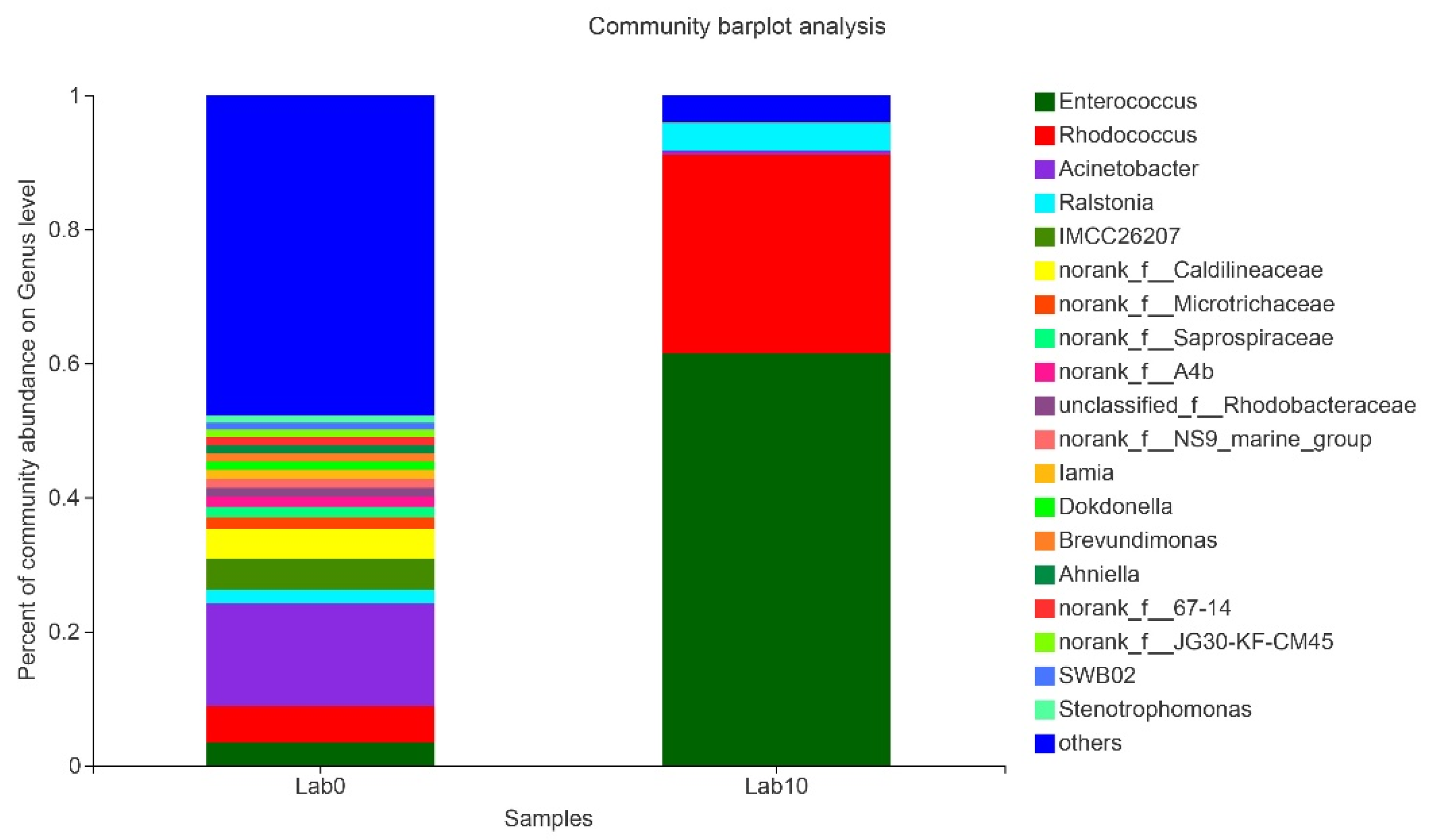
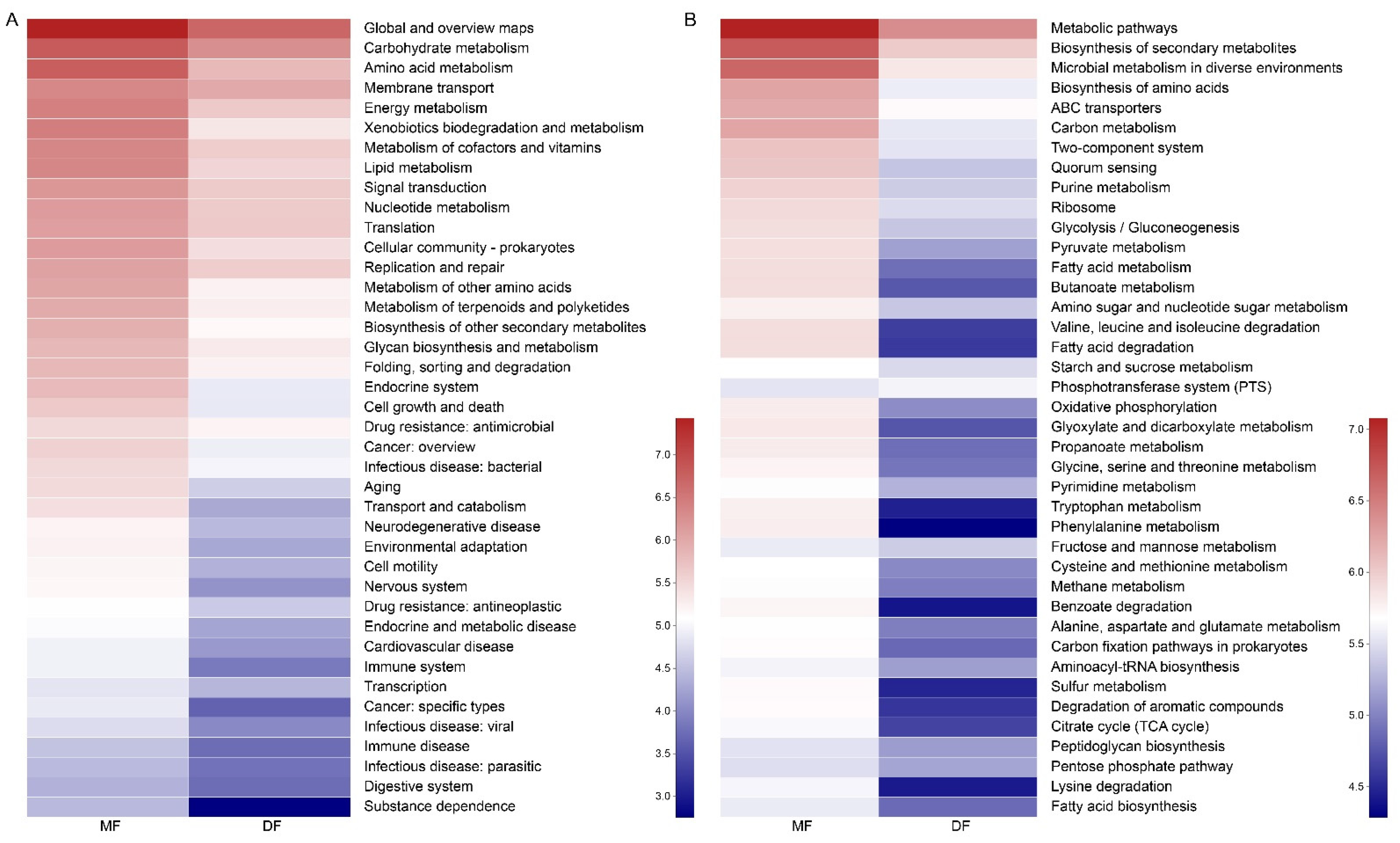
3.5. Comparison of Gut Microbiota of S. frugiperda Fed Maize and Artificial Diet
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, M.; Hulcr, J.; Sun, J.H. The role of symbiotic microbes in insect invasions. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 487–505. [Google Scholar] [CrossRef]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Vivero, R.J.; Jaramillo, N.G.; Cadavid-Restrepo, G.; Soto, S.I.; Herrera, C.X. Structural differences in gut bacteria communities in developmental stages of natural populations of Lutzomyia evansi from Colombia’s Caribbean coast. Parasites Vectors 2016, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Feldhaar, H.; Straka, J.; Krischke, M.; Berthold, K.; Stoll, S.; Mueller, M.J.; Gross, R. Nutritional upgrading for omnivorous carpenter ants by the endosymbiont Blochmannia. BMC Biol. 2007, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, G.; Elbourne, L.D.; Kinjo, Y.; Saitoh, S.; Sabree, Z.; Hojo, M.; Yamada, A.; Hayashi, Y.; Shigenobu, S.; Bandi, C.; et al. Maintenance of essential amino acid synthesis pathways in the Blattabacterium cuenoti symbiont of a wood-feeding cockroach. Biol. Lett. 2013, 9, 20121153. [Google Scholar] [CrossRef]
- Shigenobu, S.; Watanabe, H.; Hattori, M.; Sakaki, Y.; Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 2000, 407, 81–86. [Google Scholar] [CrossRef]
- McCutcheon, J.P.; Moran, N.A. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19392–19397. [Google Scholar] [CrossRef]
- Oliver, K.M.; Moran, N.A.; Hunter, M.S. Variation in resistance to parasitism in aphids is due to symbionts not host genotype. Proc. Natl. Acad. Sci. USA 2005, 102, 12795–12800. [Google Scholar] [CrossRef]
- Cardoza, Y.J.; Klepzig, K.D.; Raffa, K.F. Bacteria in oral secretions of an endophytic insect inhibit antagonistic fungi. Ecol. Entomol. 2006, 31, 636–645. [Google Scholar] [CrossRef]
- Florez, L.V.; Biedermann, P.H.; Engl, T.; Kaltenpoth, M. Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat. Prod. Rep. 2015, 32, 904–936. [Google Scholar] [CrossRef]
- Chen, B.S.; Zhang, N.; Xie, S.; Zhang, X.; He, J.; Muhammad, A.; Sun, C.; Lu, X.; Shao, Y. Gut bacteria of the silkworm Bombyx mori facilitate host resistance against the toxic effects of organophosphate insecticides. Environ. Int. 2020, 143, 105886. [Google Scholar] [CrossRef]
- Wang, G.-H.; Dittmer, J.; Douglas, B.; Huang, L.; Brucker, R.M. Coadaptation between host genome and microbiome under long-term xenobiotic-induced selection. Sci. Adv. 2021, 7, eabd4473. [Google Scholar] [CrossRef]
- Xiang, H.; Wei, G.F.; Jia, S.; Huang, J.; Miao, X.X.; Zhou, Z.; Zhao, L.-P.; Huang, Y.-P. Microbial communities in the larval midgut of laboratory and field populations of cotton bollworm (Helicoverpa armigera). Can. J. Microbiol. 2006, 52, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.S.; Currie, C.R.; Cardoza, Y.; Klepzig, K.D.; Raffa, K.F. Effects of symbiotic bacteria and tree chemistry on the growth and reproduction of bark beetle fungal symbionts. Can. J. For. Res. 2009, 39, 1133–1147. [Google Scholar] [CrossRef]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.J.; Zhang, J.L.; Zhang, R.L.; Huang, Z.D.; Wan, Q.; Zhang, Z. Comparative analysis of gut bacterial communities in housefly larvae fed different diets using a high-throughput sequencing approach. FEMS Microbiol. Lett. 2019, 366, fnz126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Shen, Z.J.; Yu, J.M.; Li, Z.; Liu, X.X.; Xu, H.L. Comparison of gut bacterial communities and their associations with host diets in four fruit borers. Pest Manag. Sci. 2020, 76, 1353–1362. [Google Scholar] [CrossRef]
- Perez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol. Ecol. 2015, 91, fiv022. [Google Scholar] [CrossRef]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host-microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef]
- Wang, W.W.; He, P.Y.; Zhang, Y.Y.; Liu, T.X.; Jing, X.F.; Zhang, S.Z. The Population Growth of Spodoptera frugiperda on Six Cash Crop Species and Implications for Its Occurrence and Damage Potential in China. Insects 2020, 11, 639. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Berasategui, A.; Salem, H.; Paetz, C.; Santoro, M.; Gershenzon, J.; Kaltenpoth, M.; Schmidt, A. Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol. Ecol. 2017, 26, 4099–4110. [Google Scholar] [CrossRef]
- Rozadilla, G.; Cabrera, N.A.; Virla, E.G.; Greco, N.M.; McCarthy, C.B. Gut microbiota of Spodoptera frugiperda (J.E. Smith) larvae as revealed by metatranscriptomic analysis. J. Appl. Entomol. 2020, 144, 351–363. [Google Scholar] [CrossRef]
- Gomes, A.F.F.; Omoto, C.; Cônsoli, F.L. Gut bacteria of field-collected larvae of Spodoptera frugiperda undergo selection and are more diverse and active in metabolizing multiple insecticides than laboratory-selected resistant strains. J. Pest Sci. 2020, 93, 833–851. [Google Scholar] [CrossRef]
- Lv, D.B.; Liu, X.Y.; Dong, Y.L.; Yan, Z.Z.; Zhang, X.; Wang, P.; Yuan, X.Q.; Li, Y.P. Comparison of gut bacterial communities of fall armyworm (Spodoptera frugiperda) reared on different host plants. Int. J. Mol. Sci. 2021, 22, 11266. [Google Scholar] [CrossRef]
- Shen, S.K.; Dowd, P.F. Detoxification spectrum of the cigarette beetle symbiont Symbiotaphrina kochii in culture. Entomol. Exp. Appl. 1991, 60, 51–59. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, X.; Chen, Z.; Wang, Z.; Lu, Y.; Cheng, D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018, 9, 114. [Google Scholar] [CrossRef]
- Aguirre, L.M.; Scully, E.D.; Trick, H.N.; Zhu, K.Y.; Smith, C.M. Comparative analyses of transcriptional responses of Dectes texanus LeConte (Coleoptera: Cerambycidae) larvae fed on three different host plants and artificial diet. Sci. Rep. 2021, 11, 11448. [Google Scholar] [CrossRef]
- Prasanna, B.M.; Huesing, J.E.; Eddy, R.; Peschke, V.M. Fall Armyworm in Africa: A Guide for Integrated Pest Management; International Maize and Wheat Improvement Center (CIMMYT): Ciudad de Mexico, Mexico, 2018; pp. 51–54. [Google Scholar]
- Chen, B.; Yu, T.; Xie, S.; Du, K.; Liang, X.; Lan, Y.; Sun, C.; Lu, X.; Shao, Y. Comparative shotgun metagenomic data of the silkworm Bombyx mori gut microbiome. Sci. Data 2018, 5, 180285. [Google Scholar] [CrossRef]
- Zhang, N.; He, J.T.; Shen, X.Q.; Sun, C.; Muhammad, A.; Shao, Y.Q. Contribution of sample processing to gut microbiome analysis in the model Lepidoptera, silkworm Bombyx mori. Comput. Struct. Biotechnol. 2021, 19, 4658–4668. [Google Scholar] [CrossRef]
- Muhammad, A.; He, J.T.; Yu, T.; Sun, C.; Shi, D.; Jiang, Y.; Xianyu, Y.; Shao, Y. Dietary exposure of copper and zinc oxides nanoparticles affect the fitness, enzyme activity, and microbial community of the model insect, silkworm Bombyx mori. Sci. Total Environ. 2022, 813, 152608. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Tan, G.C.; Wang, H.Y.; Gai, X.P. Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. Eur. J. Soil Biol. 2016, 74, 1–8. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zheng, D.; Zhong, H.; Qin, B.; Gurr, G.; Vasseur, L.; Lin, H.; Bai, J.; He, W.; You, M. DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS ONE 2013, 8, e68852. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Teh, B.S.; Sun, C.; Hu, S.R.; Lu, X.M.; Boland, W.; Shao, Y. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef]
- Snyman, M.; Gupta, A.K.; Bezuidenhout, C.C.; Claassens, S.; van den Berg, J. Gut microbiota of Busseola fusca (Lepidoptera: Noctuidae). World, J. Microbiol. Biotechnol. 2016, 32, 115. [Google Scholar] [CrossRef]
- Bapatla, K.G.; Singh, A.; Yeddula, S.; Patil, R.H. Annotation of gut bacterial taxonomic and functional diversity in Spodoptera litura and Spilosoma obliqua. J. Basic. Microbiol. 2018, 58, 217–226. [Google Scholar] [CrossRef]
- Xia, X.F.; Sun, B.T.; Gurr, G.M.; Vasseur, L.; Xue, M.Q.; You, M.S. Gut microbiota mediate insecticide resistance in the diamondback moth, Plutella xylostella (L.). Front. Microbiol. 2018, 9, 25. [Google Scholar] [CrossRef]
- Chen, B.S.; Du, K.Q.; Sun, C.; Vimalanathan, A.; Liang, X.L.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef]
- Kyi, A.; Zalucki, M.P.; Titmarsh, I.J. An experimental study of early stage survival of Helicoverpa armigera (Lepidoptera: Noctuidae) on cotton. Bull. Entomol. Res. 1991, 81, 263–271. [Google Scholar] [CrossRef]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Moll, R.M.; Romoser, W.S.; Modrzakowski, M.C.; Moncayo, A.C.; Lerdthusnee, K. Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J. Med. Entomol. 2001, 38, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Sun, J.Z.; Nguyen, H.D.; Singh, D.; Lee, K.C.; Beyenal, H.; Chen, S.-L. In-situ oxygen profiling and lignin modification in guts of wood-feeding termites. Insect Sci. 2010, 17, 277–290. [Google Scholar] [CrossRef]
- Broderick, N.A.; Raffa, K.F.; Goodman, R.M.; Handelsman, J. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 2004, 70, 293–300. [Google Scholar] [CrossRef]
- Priya, N.G.; Ojha, A.; Kajla, M.K.; Raj, A.; Rajagopal, R. Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS ONE 2012, 7, e30768. [Google Scholar] [CrossRef]
- Engel, P.; Martinson, V.G.; Moran, N.A. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. USA 2012, 109, 11002–11007. [Google Scholar] [CrossRef]
- Anand, A.A.; Vennison, S.J.; Sankar, S.G.; Prabhu, D.; Vasan, P.T.; Raghuraman, T.; Geoffrey, C.J.; Vendan, S.E. Isolation and characterization of bacteria from the gut of bombyx mori that degrade cellulose, xylan, pectin and starch and their impact on digestion. J. Insect Sci. 2010, 10, 20. [Google Scholar] [CrossRef]
- Warnecke, F.; Luginbühl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; McHardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar] [CrossRef]
- Lilburn, T.G.; Kim, K.S.; Ostrom, N.E.; Byzek, K.R.; Leadbetter, J.R.; Breznak, J.A. Nitrogen fixation by symbiotic and free-living spirochetes. Science 2001, 292, 2495–2498. [Google Scholar] [CrossRef]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef]
- Morales-Jimenez, J.; Zuniga, G.; Ramirez-Saad, H.C.; Hernandez-Rodriguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol. 2012, 64, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, N.; Martens, R.; Tebbe, C.C. Origin and diversity of metabolically active gut bacteria from laboratory-bred larvae of Manduca sexta (Sphingidae, Lepidoptera, Insecta). Appl. Environ. Microbiol. 2008, 74, 7189–7196. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Dhammi, P.; Saini, H.S.; Kaur, S. Pathogenicity of bacteria isolated from gut of Spodoptera litura (Lepidoptera: Noctuidae) and fitness costs of insect associated with consumption of bacteria. J. Invertebr. Pathol. 2015, 127, 38–46. [Google Scholar] [CrossRef]
- Vilanova, C.; Baixeras, J.; Latorre, A.; Porcar, M. The generalist inside the specialist: Gut bacterial communities of two insect species feeding on toxic plants are dominated by Enterococcus sp. Front. Microbiol. 2016, 7, 1005. [Google Scholar] [CrossRef] [PubMed]
- Egert, M.; Marhan, S.; Wagner, B.; Scheu, S.; Friedrich, M.W. Molecular profiling of 16S rRNA genes reveals diet-related differences of microbial communities in soil, gut, and casts of Lumbricus terrestris L. (Oligochaeta: Lumbricidae). FEMS Microbiol. Ecol. 2004, 48, 187–197. [Google Scholar] [CrossRef]
- Antwis, R.E.; Haworth, R.L.; Engelmoer, D.J.; Ogilvy, V.; Fidgett, A.L.; Preziosi, R.F. Ex situ diet influences the bacterial community associated with the skin of red-eyed tree frogs (Agalychnis callidryas). PLoS ONE 2014, 9, e85563. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef]
- Mason, C.J.; Clair, A.S.; Peiffer, M.; Gomez, E.; Jones, A.G.; Felton, G.W.; Hoover, K. Diet influences proliferation and stability of gut bacterial populations in herbivorous lepidopteran larvae. PLoS ONE 2020, 15, e0229848. [Google Scholar] [CrossRef]
- Erkosar, B.; Yashiro, E.; Zajitschek, F.; Friberg, U.; Maklakov, A.A.; van der Meer, J.R.; Kawecki, T.J. Host diet mediates a negative relationship between abundance and diversity of Drosophila gut microbiota. Ecol. Evol. 2018, 8, 9491–9502. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Lan, Y.; Sun, C.; Shao, Y. Insect microbial symbionts as a novel source for biotechnology. World J. Microbiol. Biotechnol. 2019, 35, 25. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, H.; Du, Y.; Idrees, A.; He, L.; Chen, J.; Ji, Q.E. Molecular identification of cultivable bacteria in the gut of adult Bactrocera tau (Walker) and their trapping effect. Pest Manag. Sci. 2018, 74, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Vacheron, J.; Pechy-Tarr, M.; Brochet, S.; Heiman, C.M.; Stojiljkovic, M.; Maurhofer, M.; Keel, C. T6SS contributes to gut microbiome invasion and killing of an herbivorous pest insect by plant-beneficial Pseudomonas protegens. ISME J. 2019, 13, 1318–1329. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.-D.; Li, J.-Y.; Hu, Z.-Q.; Liu, T.-X.; Zhang, S.-Z. Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets. Insects 2022, 13, 762. https://doi.org/10.3390/insects13090762
Li D-D, Li J-Y, Hu Z-Q, Liu T-X, Zhang S-Z. Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets. Insects. 2022; 13(9):762. https://doi.org/10.3390/insects13090762
Chicago/Turabian StyleLi, Dan-Dan, Jin-Yang Li, Zu-Qing Hu, Tong-Xian Liu, and Shi-Ze Zhang. 2022. "Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets" Insects 13, no. 9: 762. https://doi.org/10.3390/insects13090762
APA StyleLi, D.-D., Li, J.-Y., Hu, Z.-Q., Liu, T.-X., & Zhang, S.-Z. (2022). Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets. Insects, 13(9), 762. https://doi.org/10.3390/insects13090762