
Temporal Expression Profiles Reveal Potential Targets during Postembryonic Development of Forensically Important Sarcophaga peregrina (Diptera: Sarcophagidae)

 ,
,
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Rearing and Sampling of S. peregrina
2.2. RNA-Seq and Data Analysis
2.3. qRT-PCR Analysis
2.4. Statistical Analysis
3. Results
3.1. Overview of the RNA-Seq Data
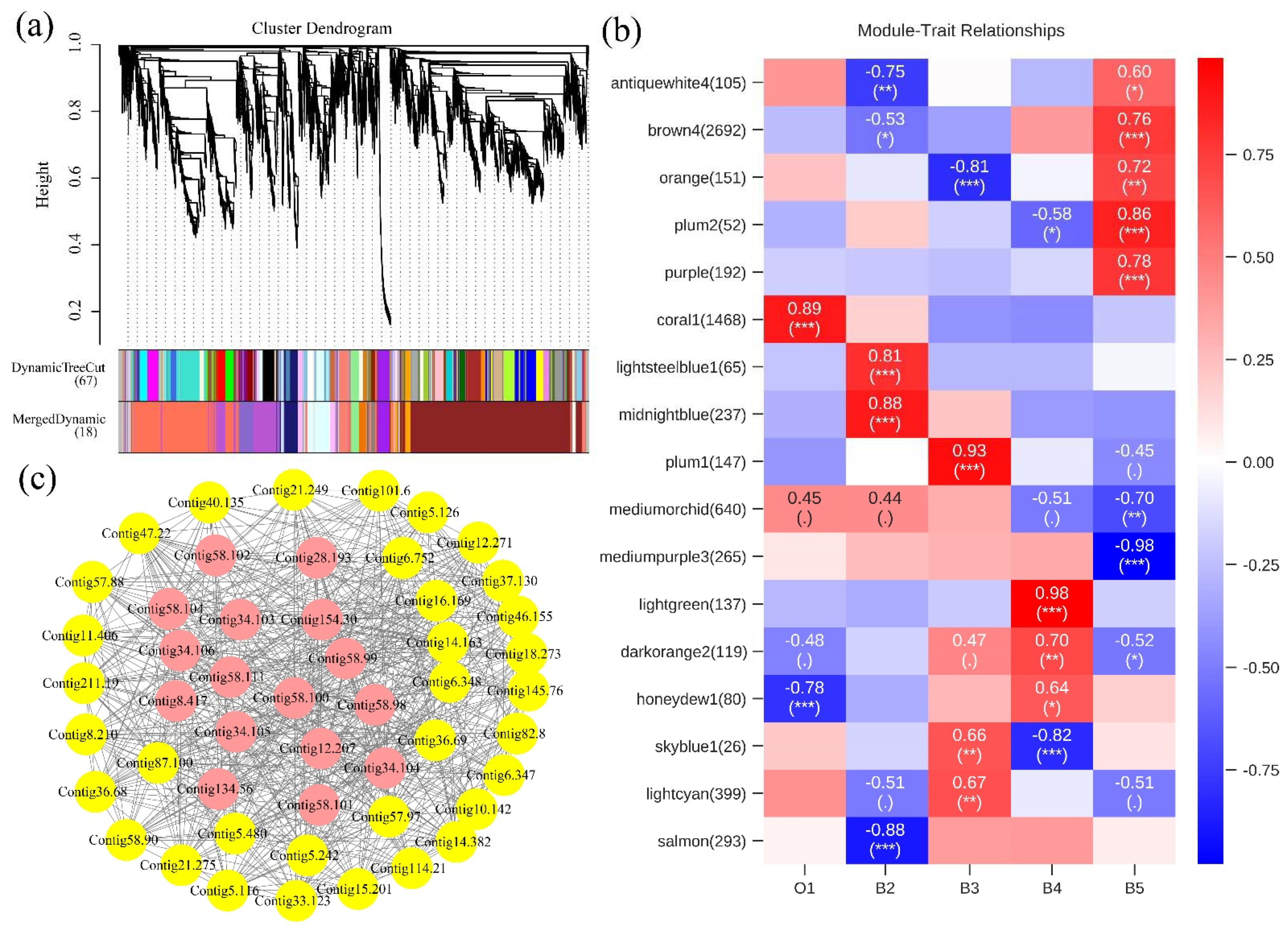
3.2. Changes in Gene Expression Profiles during Developmental Stages
3.3. Gene Functional Annotation and Enrichment Analysis
3.4. Gene Expression Level Changes in 20-Hydroxyecdysone (20E) and Juvenile Hormone (JH) Pathways
3.5. Dynamic Expression Level Changes of Genes in Chitin-Related Pathway
3.6. Dynamic Expression in Cuticle-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomberlin, J.K.; Mohr, R.; Benbow, M.E.; Tarone, A.M.; VanLaerhoven, S. A roadmap for bridging basic and applied research in forensic entomology. Annu. Rev. Entomol. 2011, 56, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Grassberger, M.; Reiter, C. Effect of temperature on Lucilia sericata (Diptera: Calliphoridae) development with special reference to the isomegalen- and isomorphen-diagram. Forensic Sci. Int. 2001, 120, 32–36. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.F.; Zhang, Y.N.; Tao, L.Y.; Wang, M. Forensically important Sarcophaga peregrina (Diptera: Sarcophagidae) in China: Development pattern and significance for estimating postmortem interval. J. Med. Entomol. 2017, 54, 1491–1497. [Google Scholar] [CrossRef]
- Baqué, M.; Filmann, N.; Verhoff, M.A.; Amendt, J. Establishment of developmental charts for the larvae of the blow fly Calliphora vicina using quantile regression. Forensic Sci. Int. 2015, 248, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Charabidze, D.; Hedouin, V. Temperature: The weak point of forensic entomology. Int J. Leg. Med. 2019, 133, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, S. Post-mortem interval estimation based on insect evidence: Current challenges. Insects 2021, 12, 314. [Google Scholar] [CrossRef] [PubMed]
- Baqué, M.; Amendt, J.; Verhoff, M.A.; Zehner, R. Descriptive analyses of differentially expressed genes during larval development of Calliphora vicina (Diptera: Calliphoridae). Int. J. Leg. Med. 2015, 129, 891–902. [Google Scholar] [CrossRef]
- Boehme, P.; Spahn, P.; Amendt, J.; Zehner, R. The analysis of temporal gene expression to estimate the age of forensically important blow fly pupae: Results from three blind studies. Int. J. Leg. Med. 2014, 128, 565–573. [Google Scholar] [CrossRef]
- Zajac, B.K.; Amendt, J.; Horres, R.; Verhoff, M.A.; Zehner, R. De novo transcriptome analysis and highly sensitive digital gene expression profiling of Calliphora vicina (Diptera: Calliphoridae) pupae using MACE (Massive Analysis of cDNA Ends). Forensic Sci. Int. Genet. 2015, 15, 137–146. [Google Scholar] [CrossRef]
- Tarone, A.M.; Foran, D.R. Gene expression during blow fly development: Improving the precision of age estimates in forensic entomology. J. Forensic Sci. 2011, 56 (Suppl. S1), S112–S122. [Google Scholar] [CrossRef]
- Pape, T. Catalogue of the Sarcophagidae of the world (Insecta: Diptera). Mem. Entomol. Int. 1996, 8, 1–558. [Google Scholar]
- Buenaventura, E.; Szpila, K.; Cassel, B.K.; Wiegmann, B.M.; Pape, T. Anchored hybrid enrichment challenges the traditional classification of flesh flies (Diptera: Sarcophagidae). Syst. Entomol. 2020, 45, 281–301. [Google Scholar] [CrossRef]
- Xue, W.Q.; Verves, Y.G.; Du, J. A review of subtribe Boettcheriscina Verves 1990 (Diptera: Sarcophagidae), with descriptions of a new species and genus from China. Ann. Soc. Ent. Fr. 2011, 47, 303–329. [Google Scholar] [CrossRef]
- Majumder, M.Z.R.; Dash, M.K.; Khan, H.R.; Khan, R.A. The reproductive biology of flesh fly, Sarcophaga peregrina (Robineau-Desvoidy, 1830) (Diptera: Sarcophagidae). Dhaka Univ. J. Biol. Sci. 2014, 23, 61–67. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Ma, M.Y.; Jiang, X.Y.; Wang, J.F.; Li, L.L.; Yin, X.J.; Wang, M.; Lai, Y.; Tao, L.Y. Insect succession on remains of human and animals in Shenzhen, China. Forensic Sci. Int. 2017, 271, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Sukontason, K.; Bunchu, N.; Chaiwong, T.; Moophayak, K.; Sukontason, K.L. Forensically important flesh fly species in Thailand: Morphology and developmental rate. Parasitol. Res. 2010, 106, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Toukairin, Y.; Arai, T.; Hoshi, T.; Oliva Trejo, J.A.; Nogami, M. The geographical distribution of fly larvae on corpses in Saitama Prefecture in Japan during the summer season. Leg. Med. 2017, 24, 75–77. [Google Scholar] [CrossRef]
- Lee, Y.T.; Chen, T.L.; Lin, Y.C.; Fung, C.P.; Cho, W.L. Nosocomial nasal myiasis in an intubated patient. J. Chin. Med. Assoc. 2011, 74, 369–371. [Google Scholar] [CrossRef]
- Miura, M.; Hayasaka, S.; Yamada, T.; Hayasaka, Y.; Kamimura, K. Ophthalmomyiasis caused by larvae of Sarcophaga peregrina. Jpn. J. Ophthalmol. 2005, 49, 177–179. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lim, H.Y.; Shin, S.E.; Cha, H.K.; Seo, J.H.; Kim, S.K.; Park, S.H.; Son, G.H. Comprehensive transcriptome analysis of Sarcophaga peregrina, a forensically important fly species. Sci. Data 2018, 5, 180220. [Google Scholar] [CrossRef]
- Shang, Y.; Ren, L.; Yang, L.; Wang, S.; Chen, W.; Dong, J.; Ma, H.; Qi, X.; Guo, Y. Differential gene expression for age estimation of forensically important Sarcophaga peregrina (Diptera: Sarcophagidae) intrapuparial. J. Med. Entomol. 2020, 57, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.H.; Ye, G.Y.; Li, K.; Hu, C.; Xu, X.H. Determining the age of adult flesh flies, Sarcophaga peregrina, using pteridine fluorescence. Med. Vet. Entomol. 2013, 27, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Rolff, J.; Johnston, P.R.; Reynolds, S. Complete metamorphosis of insects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190063. [Google Scholar] [CrossRef] [PubMed]
- Truman, J.W.; Riddiford, L.M. The origins of insect metamorphosis. Nature 1999, 401, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Nachman, R.J.; Strey, A.; Zubrzak, P.; Zdarek, J. A comparison of the pupariation acceleration activity of pyrokinin-like peptides native to the flesh fly: Which peptide represents the primary pupariation factor? Peptides 2006, 27, 527–533. [Google Scholar] [CrossRef]
- Zhu, K.Y.; Merzendorfer, H.; Zhang, W.; Zhang, J.; Muthukrishnan, S. Biosynthesis, turnover, and functions of chitin in insects. Annu. Rev. Entomol. 2016, 61, 177–196. [Google Scholar] [CrossRef]
- Santos, C.G.; Humann, F.C.; Hartfelder, K. Juvenile hormone signaling in insect oogenesis. Curr. Opin. Insect Sci. 2019, 31, 43–48. [Google Scholar] [CrossRef]
- Luo, W.; Veeran, S.; Wang, J.; Li, S.; Li, K.; Liu, S.N. Dual roles of juvenile hormone signaling during early oogenesis in Drosophila. Insect Sci. 2020, 27, 665–674. [Google Scholar] [CrossRef]
- Ren, L.; Shang, Y.; Yang, L.; Wang, S.; Wang, X.; Chen, S.; Bao, Z.; An, D.; Meng, F.; Cai, J.; et al. Chromosome-level de novo genome assembly of Sarcophaga peregrina provides insights into the evolutionary adaptation of flesh flies. Mol. Ecol. Resour. 2021, 21, 251–262. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Young, A.; Whitehouse, N.; Cho, J.; Shaw, C. OntologyTraverser: An R package for GO analysis. Bioinformatics 2005, 21, 275–276. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Merkey, A.B.; Wong, C.K.; Hoshizaki, D.K.; Gibbs, A.G. Energetics of metamorphosis in Drosophila melanogaster. J. Insect Physiol. 2011, 57, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Merzendorfer, H.; Zimoch, L. Chitin metabolism in insects: Structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef] [PubMed]
- Grau, V.; Lafont, R. Metabolism of ecdysone and 20-hydroxyecdysone in adult Drosophila melanogaster. Insect Biochem. Mol. Biol. 1994, 24, 49–58. [Google Scholar] [CrossRef]
- Chen, E.H.; Hou, Q.L.; Wei, D.D.; Dou, W.; Liu, Z.; Yang, P.J.; Smagghe, G.; Wang, J.J. Tyrosine hydroxylase coordinates larval-pupal tanning and immunity in oriental fruit fly (Bactrocera dorsalis). Pest Manag. Sci. 2018, 74, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Arbeitman, M.N.; Furlong, E.E.; Imam, F.; Johnson, E.; Null, B.H.; Baker, B.S.; Krasnow, M.A.; Scott, M.P.; Davis, R.W.; White, K.P. Gene expression during the life cycle of Drosophila melanogaster. Science 2002, 297, 2270–2275. [Google Scholar] [CrossRef]
- Fremdt, H.; Amendt, J.; Zehner, R. Diapause-specific gene expression in Calliphora vicina (Diptera: Calliphoridae)—A useful diagnostic tool for forensic entomology. Int. J. Leg. Med. 2014, 128, 1001–1011. [Google Scholar] [CrossRef]
- Boehme, P.; Spahn, P.; Amendt, J.; Zehner, R. Differential gene expression during metamorphosis: A promising approach for age estimation of forensically important Calliphora vicina pupae (Diptera: Calliphoridae). Int. J. Leg. Med. 2013, 127, 243–249. [Google Scholar] [CrossRef]
- Vincent, J.F.; Wegst, U.G. Design and mechanical properties of insect cuticle. Arthropod. Struct. Dev. 2004, 33, 187–199. [Google Scholar] [CrossRef]
- Shukla, E.; Thorat, L.J.; Nath, B.B.; Gaikwad, S.M. Insect trehalase: Physiological significance and potential applications. Glycobiology 2015, 25, 357–367. [Google Scholar] [CrossRef]
- Arakane, Y.; Dixit, R.; Begum, K.; Park, Y.; Specht, C.A.; Merzendorfer, H.; Kramer, K.J.; Muthukrishnan, S.; Beeman, R.W. Analysis of functions of the chitin deacetylase gene family in Tribolium castaneum. Insect Biochem. Mol. Biol. 2009, 39, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Huang, L.X.; Gong, Y.J.; Zheng, S.C.; Liu, L.; Huang, L.H.; Feng, Q.L. De novo characterization of transcriptome and gene expression dynamics in epidermis during the larval-pupal metamorphosis of common cutworm. Insect Biochem. Mol. Biol. 2013, 43, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.H.; Hou, Q.L.; Dou, W.; Wei, D.D.; Yue, Y.; Yang, R.L.; Yu, S.F.; De Schutter, K.; Smagghe, G.; Wang, J.J. RNA-seq analysis of gene expression changes during pupariation in Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). BMC Genom. 2018, 19, 693. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Xia, Y.D.; Zhang, Q.; Li, W.; Li, R.Y.; Liu, Y.; Chen, E.H.; Dou, W.; Stelinski, L.L.; Wang, J.J. Potential targets for controlling Bactrocera dorsalis using cuticle- and hormone-related genes revealed by a developmental transcriptome analysis. Pest Manag. Sci. 2020, 76, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Bellés, X.; Martín, D.; Piulachs, M.D. The mevalonate pathway and the synthesis of juvenile hormone in insects. Annu. Rev. Entomol. 2005, 50, 181–199. [Google Scholar] [CrossRef]
- Riddiford, L.M.; Truman, J.W.; Mirth, C.K.; Shen, Y.C. A role for juvenile hormone in the prepupal development of Drosophila melanogaster. Development 2010, 137, 1117–1126. [Google Scholar] [CrossRef]
- Petryk, A.; Warren, J.T.; Marqués, G.; Jarcho, M.P.; Gilbert, L.I.; Kahler, J.; Parvy, J.P.; Li, Y.; Dauphin-Villemant, C.; O’Connor, M.B. Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. Proc. Natl. Acad. Sci. USA 2003, 100, 13773–13778. [Google Scholar] [CrossRef]
- Cabrera, A.R.; Shirk, P.D.; Evans, J.D.; Hung, K.; Sims, J.; Alborn, H.; Teal, P.E. Three Halloween genes from the Varroa mite, Varroa destructor (Anderson & Trueman) and their expression during reproduction. Insect Mol. Biol. 2015, 24, 277–292. [Google Scholar] [CrossRef]
- Sugahara, R.; Tanaka, S.; Shiotsuki, T. RNAi-mediated knockdown of SPOOK reduces ecdysteroid titers and causes precocious metamorphosis in the desert locust Schistocerca gregaria. Dev. Biol. 2017, 429, 71–80. [Google Scholar] [CrossRef]
- Jindra, M.; Bittova, L. The juvenile hormone receptor as a target of juvenoid “insect growth regulators”. Arch. Insect Biochem. Physiol. 2020, 103, 21615. [Google Scholar] [CrossRef]
- Jindra, M.; Palli, S.R.; Riddiford, L.M. The juvenile hormone signaling pathway in insect development. Annu. Rev. Entomol. 2013, 58, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Jia, Q.Q.; Li, S. Juvenile hormone signaling—A mini review. Insect Sci. 2019, 26, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Qiu, Y.W.; Huang, J.; Tobe, S.S.; Chen, S.S.; Kai, Z.P. Enzymes in the juvenile hormone biosynthetic pathway can be potential targets for pest control. Pest Manag. Sci. 2020, 76, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, M.E. Control of oxidative phosphorylation during insect metamorphosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, 314–321. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Wrońska, A.K.; Kazek, M.; Boguś, M.I. Metamorphosis-related changes in the free fatty acid profiles of Sarcophaga (Liopygia) argyrostoma (Robineau-Desvoidy, 1830). Sci. Rep. 2020, 10, 17337. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, L.; Shang, Y.; Zhang, X.; Chen, S.; Zheng, Y.; Zou, Y.; Qu, Y.; Cai, J.; Zhang, C.; Guo, Y. Temporal Expression Profiles Reveal Potential Targets during Postembryonic Development of Forensically Important Sarcophaga peregrina (Diptera: Sarcophagidae). Insects 2022, 13, 453. https://doi.org/10.3390/insects13050453
Ren L, Shang Y, Zhang X, Chen S, Zheng Y, Zou Y, Qu Y, Cai J, Zhang C, Guo Y. Temporal Expression Profiles Reveal Potential Targets during Postembryonic Development of Forensically Important Sarcophaga peregrina (Diptera: Sarcophagidae). Insects. 2022; 13(5):453. https://doi.org/10.3390/insects13050453
Chicago/Turabian StyleRen, Lipin, Yanjie Shang, Xiangyan Zhang, Shan Chen, Yunna Zheng, Ying Zou, Yihong Qu, Jifeng Cai, Changquan Zhang, and Yadong Guo. 2022. "Temporal Expression Profiles Reveal Potential Targets during Postembryonic Development of Forensically Important Sarcophaga peregrina (Diptera: Sarcophagidae)" Insects 13, no. 5: 453. https://doi.org/10.3390/insects13050453
APA StyleRen, L., Shang, Y., Zhang, X., Chen, S., Zheng, Y., Zou, Y., Qu, Y., Cai, J., Zhang, C., & Guo, Y. (2022). Temporal Expression Profiles Reveal Potential Targets during Postembryonic Development of Forensically Important Sarcophaga peregrina (Diptera: Sarcophagidae). Insects, 13(5), 453. https://doi.org/10.3390/insects13050453