
Adaptation of Fig Wasps (Agaodinae) to Their Host Revealed by Large-Scale Transcriptomic Data

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. mRNA-Seq Library Construction, Illumina Sequencing, Assembly, and Annotation
2.3. Ortholog Identification and Alignment
2.4. Phylogenetic Tree and Divergence Time
2.5. Gene Family Expansion and Contraction
2.6. Evolutionary Rate and Positive Selection Analyses
2.7. Annotation of Chemosensory Genes
3. Results
3.1. Comparison of Transcriptome Sequencing and Assembly among 25 Fig Wasp Species
3.2. Unigenes Annotated against Public Databases
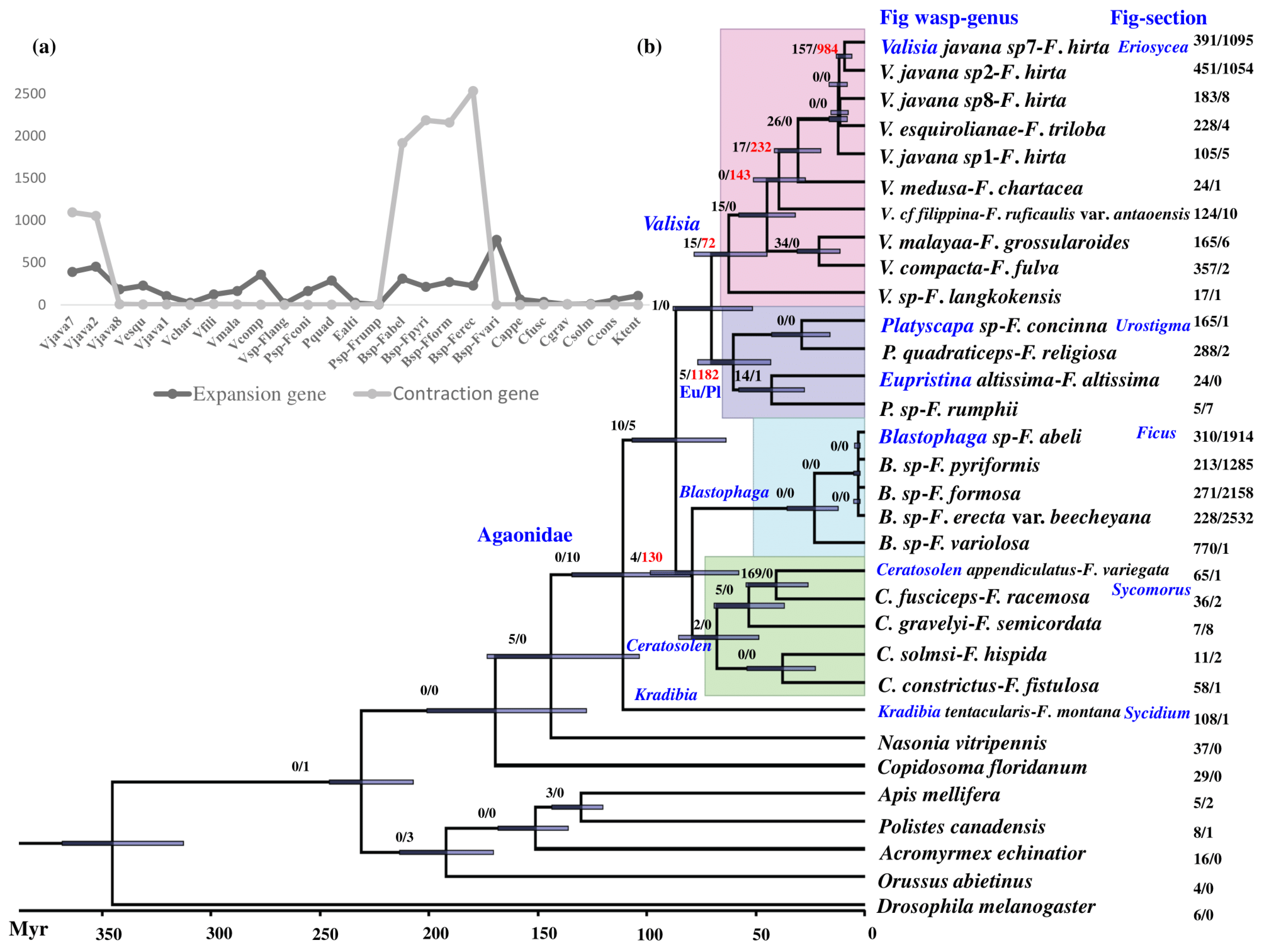
3.3. Phylogenetic Relationships
3.4. Gene Family Expansion and Contraction at the Level of Expression
3.5. Contraction of Genes Involved in Chemosensory
3.6. Rapidly Evolving Genes (REGs)
3.7. Positively Selected Genes (PSGs)
4. Discussion
4.1. Comparisons among Agaonidae
4.2. Comparisons at Genus Level
4.3. Comparisons among Species
4.4. Comparisons among Closely Related Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Melián, C.J.; Matthews, B.; Andreazzi, C.; Rodriguez, J.P.; Harmon, L.; Fortuna, M. Deciphering the Interdependence between Ecological and Evolutionary Networks. Trends Ecol. Evol. 2018, 33, 504–512. [Google Scholar] [CrossRef]
- Jazen, D.H. When is it coevolution? Evolution 1980, 34, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; Heidel-Fischer, H.M.; Bekaert, M.; Rota, J.; Glöckner, G.; Platts, A.; Heckel, D.; Der, J.P.; Wafula, E.K.; Tang, M.; et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA 2015, 112, 8362–8366. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.P.R.; Thompson, J.N. Evolution and coevolution in mutualistic networks. Ecology 2011, 14, 877–885. [Google Scholar] [CrossRef]
- Kirkness, E.F.; Haas, B.J.; Sun, W.; Braig, H.R.; Perotti, M.A.; Clark, J.M.; Lee, S.H.; Robertson, H.M.; Kennedy, R.C.; Elhaik, E.; et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. USA 2010, 107, 12168–12173. [Google Scholar] [CrossRef]
- Xiao, J.-H.; Yue, Z.; Jia, L.-Y.; Yang, X.-H.; Niu, L.-M.; Wang, Z.; Zhang, P.; Sun, B.-F.; He, S.-M.; Li, Z.; et al. Obligate mutualism within a host drives the extreme specialization of a fig wasp genome. Genome Biol. 2013, 14, R141. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhou, X.; Zhang, C.-X.; Yu, L.-L.; Fan, H.-W.; Wang, Z.; Xu, H.-J.; Xi, Y.; Zhu, Z.-R.; Zhou, W.-W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef]
- Berg, C. Flora Malesiana Precursor for the Treatment of Moraceae 1: The Main Subdivision of Ficus: The Subgenera. Blumea Biodivers. Evol. Biogeogr. Plants 2003, 48, 167–168. [Google Scholar] [CrossRef]
- Yu, H.; Tian, E.; Zheng, L.; Deng, X.; Cheng, Y.; Chen, L.; Wu, W.; Tanming, W.; Zhang, D.; Compton, S.G.; et al. Multiple parapatric pollinators have radiated across a continental fig tree displaying clinal genetic variation. Mol. Ecol. 2019, 28, 2391–2405. [Google Scholar] [CrossRef]
- Galil, J.; Eisikowitch, D. Flowering cycles and fruit types of Ficus sycomorus in Israel. N. Phytol. 1968, 67, 745–758. [Google Scholar] [CrossRef]
- Janzen, D.H. How to be a fig. Annu. Rev. Ecol. Syst. 1979, 10, 13–51. [Google Scholar] [CrossRef]
- Weiblen, G.D. How to be a fig wasp. Annu. Rev. Entomol. 2002, 47, 299–330. [Google Scholar] [CrossRef]
- Rønsted, N.; Weiblen, G.D.; Cook, J.; Salamin, N.; Machado, C.A.; Savolainen, V. 60 million years of co-divergence in the fig–wasp symbiosis. Proc. R. Soc. B Boil. Sci. 2005, 272, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Ware, A.B.; Kaye, P.T.; Compton, S.G.; Van Noort, S. Fig volatiles: Their role in attracting pollinators and maintaining pollinator specificity. Plant Syst. Evol. 1993, 186, 147–156. [Google Scholar] [CrossRef]
- Hossaert-Mckey, M.; Soler, C.; Schatz, B.; Proffit, M. Floral scents: Their roles in nursery pollination mutualisms. Chemoecology 2010, 20, 75–88. [Google Scholar] [CrossRef]
- Chen, C.; Song, Q.; Proffit, M.; Bessière, J.-M.; Li, Z.; Hossaert-Mckey, M. Private channel: A single unusual compound assures specific pollinator attraction in Ficus semicordata. Funct. Ecol. 2009, 23, 941–950. [Google Scholar] [CrossRef]
- Frank, S.A. The behavior and morphology of the fig wasps Pegoscapus assuetus and P. jimenezi: Descriptions and suggested behavioral characters for phylogenetic studies. Psyche 1984, 91, 289–308. [Google Scholar] [CrossRef][Green Version]
- Verkerke, W. Structure and function of the fig. Cell. Mol. Life Sci. 1989, 45, 612–622. [Google Scholar] [CrossRef]
- Ware, A.B.; Compton, S.G. Dispersal of adult female fig wasps. Èntomol. Exp. Appl. 1994, 73, 221–229. [Google Scholar] [CrossRef]
- Rose, H.A. The relationship between feeding specialization and host plants to aldrin epoxidase activities of midgut homogenates in larval Lepidoptera. Ecol. Èntomol. 1985, 10, 455–467. [Google Scholar] [CrossRef]
- Frank, M.R.; Fogleman, J.C. Involvement of cytochrome P450 in host-plant utilization by Sonoran Desert Drosophila. Proc. Natl. Acad. Sci. USA 1992, 89, 11998–12002. [Google Scholar] [CrossRef]
- Berenbaum, M.R. Postgenomic chemical ecology: From genetic code to ecological interactions. J. Chem. Ecol. 2002, 28, 873–896. [Google Scholar] [CrossRef]
- Li, W.; Schuler, M.A.; Berenbaum, M.R. Diversification of furanocoumarin-metabolizing cytochrome P450 monooxygenases in two papilionids: Specificity and substrate encounter rate. Proc. Natl. Acad. Sci. USA 2003, 100, 14593–14598. [Google Scholar] [CrossRef]
- Weiblen, G.D. Correlated Evolution in Fig Pollination. Syst. Biol. 2004, 53, 128–139. [Google Scholar] [CrossRef]
- Ramirez, W.B. Coevolution of Ficus and Agaonidae. Ann. Missorri. Bot. Gard. 1974, 61, 770–780. [Google Scholar]
- Wiebes, J.T. Co-evolution of figs and their insect pollinators. Ann. Rev. Ecol. Sys. 1979, 10, 1–12. [Google Scholar] [CrossRef]
- Berg, C.C. Reproduction and Evolution in Ficus (Moraceae): Traits connected with the adequate rearing of pollinators. Mem. N. Y. Bot. Gard. 1990, 55, 169–185. [Google Scholar]
- Cruaud, A.; Rønsted, N.; Chantarasuwan, B.; Chou, L.-S.; Clement, W.L.; Couloux, A.; Cousins, B.; Genson, G.; Harrison, R.; Hanson, P.E.; et al. An Extreme Case of Plant–Insect Codiversification: Figs and Fig-Pollinating Wasps. Syst. Biol. 2012, 61, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Peng, Y.Q.; Harder, L.D.; Huang, J.F.; Yang, D.R.; Zhang, D.Y.; Liao, W. The nature of interspecific interactions and co-diversification patterns, as illustrated by the fig microcosm. N. Phytol. 2019, 224, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.C.; Wiebes, J.T. African fig trees and fig wasps. Verh. K. Ned. Akad. Wet. Afd. Natuurkd. Tweede Reeks 1992, 89, 1–298. [Google Scholar]
- Van Noort, S.; Compton, S.G. Convergent evolution of agaonine and sycoecine (Agaonidae, Chalcidoidea) head shape in response to the constraints of host fig morphology. J. Biogeogr. 1996, 23, 415–424. [Google Scholar] [CrossRef]
- Wang, N.X.; Niu, L.M.; Bian, S.N.; Xiao, J.H.; Huang, D.W. Odorant-binding protein (OBP) genes affect host specificity in a fig-pollinator mutualistic system. Insect Mol. Biol. 2014, 23, 621–631. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Zhang, S.; Chen, S.; Wang, Y.; Wen, P.; Ma, X.; Shi, Y.; Qi, R.; Yang, Y.; et al. Genomes of the Banyan Tree and Pollinator Wasp Provide Insights into Fig-Wasp Coevolution. Cell 2020, 183, 875–889.e17. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Yu, H.; Kjellberg, F. Transcriptome analysis of genes involved in the response of a pollinator fig wasp to volatile organic compounds from its host figs. Acta Oecologica 2018, 90, 91–98. [Google Scholar] [CrossRef]
- Hill, D.S. Figs (Ficus spp.) of Hong Kong; Oxford University Press: Hong Kong, China; London, UK, 1967; pp. 1–128. [Google Scholar]
- Kusumi, J.; Azuma, H.; Tzeng, H.-Y.; Chou, L.-S.; Peng, Y.-Q.; Nakamura, K.; Su, Z.-H. Phylogenetic analyses suggest a hybrid origin of the figs (Moraceae: Ficus) that are endemic to the Ogasawara (Bonin) Islands, Japan. Mol. Phylogenetics Evol. 2012, 63, 168–179. [Google Scholar] [CrossRef]
- Wiebes, J.T. Agaonidae (Hymenoptera Chalcidoidea) and Ficus (Moraceae): Fig wasps and their figs, xi (Blastophaga) s.l. Proc. Kon. Ned. Akad. Wet. Ser. C 1993, 96, 347–367. [Google Scholar]
- Wachi, N.; Kusumi, J.; Tzeng, H.-Y.; Su, Z.-H. Genome-wide sequence data suggest the possibility of pollinator sharing by host shift in dioecious figs (Moraceae, Ficus). Mol. Ecol. 2016, 25, 5732–5746. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.H.; Sasaki, A.; Kusimi, J.; Chou, P.A.; Tzeng, H.Y.; Li, H.Q.; Yu, H. Pollinator sharing and co-pollinator, host-shift and specificity reestablishment among closely related dioecious fig species (Ficus, Moraceae). Nat. Ecol. Evol. submitted in 2021.
- Murray, M.; Thompson, W. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.; A Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; pp. 227–245. [Google Scholar]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 46, D8–D13. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; A Salazar, G.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35 (Suppl. 2), W182–W185. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Chen, F.; Mackey, A.J.; Stoeckert, C.J.; Roos, D.S. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006, 34 (Suppl. 1), D363–D368. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; DeMuth, J.P.; Hahn, M. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Zhang, Z.; He, S. Comprehensive Transcriptome Analysis Reveals Accelerated Genic Evolution in a Tibet Fish, Gymnodiptychus pachycheilus. Genome Biol. Evol. 2014, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.; Zhou, K.; Zhang, Y.; Song, Z.; He, S. Evidence for Adaptation to the Tibetan Plateau Inferred from Tibetan Loach Transcriptomes. Genome Biol. Evol. 2015, 7, 2970–2982. [Google Scholar] [CrossRef]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes Empirical Bayes Inference of Amino Acid Sites Under Positive Selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Kaupp UB: Olfactory signalling in vertebrates and insects: Differences and commonalities. Nat. Rev. Neurosci. 2010, 11, 188–200. [CrossRef]
- Wang, G.; Zhang, X.; Herre, E.A.; McKey, D.; Machado, C.A.; Yu, W.-B.; Cannon, C.H.; Arnold, M.L.; Pereira, R.A.S.; Ming, R.; et al. Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 2021, 12, 718. [Google Scholar] [CrossRef]
- Wang, R.; Yang, Y.; Jing, Y.; Segar, S.T.; Zhang, Y.; Wang, G.; Chen, J.; Liu, Q.-F.; Chen, S.; Chen, Y.; et al. Molecular mechanisms of mutualistic and antagonistic interactions in a plant–pollinator association. Nat. Ecol. Evol. 2021, 974–986. [Google Scholar] [CrossRef]
- Vieira, F.G.; Rozas, J. Comparative Genomics of the Odorant-Binding and Chemosensory Protein Gene Families across the Arthropoda: Origin and Evolutionary History of the Chemosensory System. Genome Biol. Evol. 2011, 3, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Gracia, A.; Vieira, F.G.; Rozas, J. Molecular evolution of the major chemosensory gene families in insects. Heredity 2009, 103, 208–216. [Google Scholar] [CrossRef]
- Abuin, L.; Bargeton, B.; Ulbrich, M.H.; Isacoff, E.; Kellenberger, S.; Benton, R. Functional Architecture of Olfactory Ionotropic Glutamate Receptors. Neuron 2011, 69, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-Y.; Machado, C.A.; Dang, X.; Peng, Y.; Yang, D.; Zhang, D.; Liao, W. The incidence and pattern of copollinator diversification in dioecious and monoecious figs. Evolution 2015, 69, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Tanji, T.; Ip, Y.T. Regulators of the Toll and Imd pathways in the Drosophila innate immune response. Trends Immunol. 2005, 26, 193–198. [Google Scholar] [CrossRef]
- Webb, B.A.; Strand, M.R.; Dickey, S.E.; Beck, M.H.; Hilgarth, R.S.; Barney, W.E.; Kadash, K.; Kroemer, J.A.; Lindstrom, K.G.; Rattanadechakul, W.; et al. Polydnavirus genomes reflect their dual roles as mutualists and pathogens. Virology 2006, 347, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Fath-Goodin, A.; Kroemer, J.; Martin, S.; Reeves, K.; Webb, B.A. Polydnavirus Genes that Enhance the Baculovirus Expression Vector System. Adv. Virus Res. 2006, 68, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Falabella, P.; Varricchio, P.; Provost, B.; Espagne, E.; Ferrarese, R.; Grimaldi, A.; Eguileor, M.D.; Fimiani, G.; Ursini, M.V.; Malva, C.; et al. Characterization of the IkappaBlike gene family in polydnaviruses associated with wasps belonging to different Braconid subfamilies. J. Gen. Virol. 2007, 88, 92–104. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Fig Wasp | Host | Sampling Site | ||||
|---|---|---|---|---|---|---|
| Genus | Species | Species | Section | Site | Latitude | Longitude |
| Platyscapa | quadraticeps | F. religiosa | Urotigma | Myanmar | 22.013 | 96.073 |
| sp. | F. concinna | Urotigma | China—Guangdong | 23.816 | 114.013 | |
| sp. | F. rumphii | Urotigma | Myanmar | 21.984 | 96.060 | |
| Eupristina | altissima | F. altissima | Urotigma | China—Guangdong | 22.763 | 113.612 |
| Valisia | sp. 1 | F. hirta | Eriosycea | China—SCBG | 23.179 | 113.352 |
| sp. 2 | F. hirta | Eriosycea | Thailand—Chiang Mai | 18.809 | 98.914 | |
| sp. 7 | F. hirta | Eriosycea | Thailand—Chantaburi | 12.774 | 102.096 | |
| sp. 8 | F. hirta | Eriosycea | Thailand—Trang | 7.467 | 99.639 | |
| esquirolianae | F. triloba | Eriosycea | China—Guangdong | 23.635 | 113.770 | |
| medusa | F. chartacea | Eriosycea | Thailand | 7.556 | 99.766 | |
| cf. filippina | F. ruficaulis var. antaoensis | Eriosycea | Taiwan | 21.962 | 120.811 | |
| malayana | F. grossularoides | Eriosycea | Thailand—Narathiwat | 5.799 | 101.762 | |
| compacta | F. fulva | Eriosycea | China—Guangdong | 8.776 | 99.724 | |
| sp. | F. langkokensis | Eriosycea | China—Guangdong | 24.252 | 112.036 | |
| Blastophaga | sp. | F. abeli | Ficus | China—Guangdong | 23.636 | 113.780 |
| sp. | F. pyriformis | Ficus | Thailand | 18.504 | 98.665 | |
| sp. | F. erecta var. beecheyana | Ficus | China—Guangdong | 23.761 | 113.920 | |
| sp. | F. formosa | Ficus | China—Guangdong | 23.623 | 113.811 | |
| sp. | F. variolosa | Ficus | China—Guangdong | 23.180 | 113.275 | |
| Ceratosolen | appendiculatus | F. variegata | Sycomorus | China—Guangdong | 23.181 | 113.359 |
| fusciceps | F. racemosa | Sycomorus | Myanmar | 20.765 | 96.945 | |
| gravelyi | F. semicordata | Sycomorus | Thailand—Chiang Mai | 19.362 | 98.922 | |
| constrictus | F. fistulosa | Sycomorus | China—Guangdong | 23.156 | 112.511 | |
| solmi | F. hispida | Sycomorus | China—SCBG | 23.179 | 113.352 | |
| Kradibia | tentacularis | F. montana | Sycidium | Thailand | 7.557 | 99.776 |
| Items | Gene Family | REG | PSG | |||||
|---|---|---|---|---|---|---|---|---|
| Expansion | Contraction | |||||||
| Total Number | Number of Gene Families Enriched in | Total Number | Number of Gene Families Enriched in | Total Number | Number of Genes Enriched in | Total Number | Number of Genes Enriched in | |
| GO/KEGG | GO/KEGG | GO/KEGG | GO/KEGG | |||||
| Family | ||||||||
| Agaonidae | 0 | 0/0 | 10 | 0/2 | 857 | 484/45 | 68 | 0/0 |
| Genus | ||||||||
| Eu/Pl | 5 | 0/0 | 1182 | 12/8 | 2572 | 14/0 | 85 | 13/0 |
| Ceratosolen | 2 | 0/0 | 0 | 0/0 | 2073 | 4/0 | 81 | 11/1 |
| Valisia | 15 | 0/5 | 72 | 0/0 | 2133 | 17/0 | 75 | 9/2 |
| Blastophaga | 0 | 0/0 | 0 | 0/0 | 2273 | 1/0 | 110 | 0/2 |
| Species in Eu/Pl clade | ||||||||
| Platyscapa quadraticeps | 288 | 15/0 | 2 | 0/0 | 890 | 0/5 | 23 | 0/0 |
| P. sp.-F. concinna | 165 | 20/0 | 1 | 0/0 | 1195 | 1/2 | 39 | 0/0 |
| P. sp.-F. rumphill | 5 | 0/0 | 7 | 0/0 | 1027 | 0/0 | 28 | 5/0 |
| Eupristina altissima | 24 | 0/2 | 0 | 0/0 | 1258 | 2/1 | 22 | 0/0 |
| Species in Ceratosolen | ||||||||
| C. appendiculatus | 65 | 0/14 | 1 | 0/0 | 1088 | 0/0 | 32 | 0/0 |
| C. fusciceps | 36 | 2/1 | 2 | 0/0 | 1223 | 1/2 | 39 | 0/0 |
| C. gravelyi | 7 | 0/0 | 8 | 0/0 | 1245 | 0/0 | 29 | 0/0 |
| C. solmsi | 11 | 0/0 | 2 | 0/0 | 1012 | 0/0 | 24 | 0/0 |
| C. constrictus | 58 | 77/2 | 1 | 0/0 | 1144 | 0/0 | 27 | 1/0 |
| Species in Kradibia | ||||||||
| K. tentacularis | 108 | 88/4 | 1 | 0/0 | 1649 | 0/0 | 43 | 0/0 |
| Species in Valisia | ||||||||
| V. medusa | 24 | 0/0 | 1 | 0/0 | 1864 | 4/0 | 63 | 0/0 |
| V. cf filippina | 124 | 31/8 | 10 | 0/0 | 1664 | 0/0 | 96 | 0/0 |
| V. malayana | 165 | 2/0 | 6 | 0/0 | 1242 | 0/0 | 28 | 0/0 |
| V. compacta | 357 | 75/9 | 2 | 0/0 | 1549 | 0/0 | 44 | 0/0 |
| V. sp-F. langkokensis | 17 | 0/1 | 1 | 0/0 | 1773 | 7/4 | 91 | 18/2 |
| Related species of V. javana | ||||||||
| V. javana sp. 7 | 391 | 10/4 | 1095 | 257/18 | 804 | 0/0 | 207 | 0/0 |
| V. javana sp. 2 | 451 | 79/16 | 1054 | 29/5 | 803 | 0/2 | 177 | 0/0 |
| V. javana sp. 8 | 183 | 1/0 | 8 | 0/0 | 1065 | 0/0 | 128 | 0/0 |
| V. javana sp. 1 | 105 | 4/1 | 5 | 0/0 | 1162 | 0/1 | 177 | 0/0 |
| V. esquirolianae | 228 | 16/17 | 4 | 0/0 | 1032 | 0/3 | 155 | 18/0 |
| Species in Blastophaga | ||||||||
| Related taxa—five taxa of one species in different hosts | ||||||||
| B. sp.-F. abeli | 310 | 62/1 | 1914 | 137/16 | 533 | 193/20 | 275 | 3/0 |
| B. sp.-F. pyriformis | 213 | 44/2 | 2185 | 329/31 | 967 | 112/10 | 288 | 0/0 |
| B. sp.-F. formosa | 271 | 14/3 | 2158 | 283/25 | 286 | 0/0 | 227 | 0/0 |
| B. sp.-F. erecta var. beecheyana | 228 | 31/12 | 2532 | 546/12 | 324 | 0/1 | 230 | 0/1 |
| Species | OBP | Or | CSP | Ir | Gr |
|---|---|---|---|---|---|
| Valisia javana sp. 7 | 31 | 33 | 38 | 11 | 8 |
| V. javana sp. 2 | 23 | 45 | 26 | 17 | 7 |
| V. javana sp. 8 | 16 | 27 | 16 | 15 | 9 |
| V. esquirolianae | 13 | 38 | 14 | 14 | 10 |
| V. javana sp. 1 | 15 | 30 | 16 | 16 | 6 |
| V. medusa | 19 | 30 | 24 | 11 | 8 |
| V. cf filippina | 11 | 21 | 24 | 10 | 9 |
| V. malayana | 16 | 21 | 28 | 7 | 6 |
| V. compacta | 13 | 21 | 14 | 13 | 9 |
| V. sp.-F. langkokensis | 16 | 20 | 23 | 17 | 6 |
| Platyscapa quadraticeps | 11 | 26 | 16 | 11 | 10 |
| P. sp.-F. concinna | 8 | 23 | 23 | 6 | 5 |
| P. sp.-F. rumphill | 6 | 20 | 15 | 10 | 9 |
| Eupristina altissima | 7 | 73 | 16 | 10 | 11 |
| Blastophaga sp.-F. abeli | 11 | 22 | 20 | 13 | 15 |
| B. sp.-F. pyriformis | 11 | 20 | 13 | 13 | 10 |
| B. sp.-F. formosa | 9 | 27 | 14 | 14 | 14 |
| B. sp.-F. erecta var. beecheyana | 12 | 30 | 14 | 17 | 15 |
| B. sp.-F. variolosa | 29 | 27 | 24 | 11 | 12 |
| Ceratosolen appendiculatus | 14 | 78 | 19 | 22 | 19 |
| C. fusciceps | 10 | 63 | 16 | 18 | 15 |
| C. gravelyi | 11 | 43 | 12 | 11 | 9 |
| C. solmsi | 9 | 47 | 14 | 14 | 12 |
| C. constrictus | 12 | 34 | 16 | 14 | 6 |
| Kradibia tentacularis | 35 | 22 | 32 | 14 | 15 |
| Mean for 25 fig wasps | 14.72 | 33.64 | 19.48 | 13.16 | 10.2 |
| SE | 7.45 | 16.39 | 6.59 | 3.56 | 3.63 |
| Nasonia vitripennis | 62 | 474 | 20 | 53 | 143 |
| Copidosoma floridanum | 47 | 186 | 16 | 38 | 57 |
| Apis mellifera | 21 | 343 | 13 | 31 | 68 |
| Polistes canadensis | 11 | 181 | 18 | 29 | 48 |
| Acromyrmex echinatior | 17 | 681 | 31 | 38 | 93 |
| Orussus abietinus | 9 | 94 | 26 | 31 | 39 |
| Drosophila melanogaster | 48 | 129 | 9 | 60 | 82 |
| Mean for seven other insects | 30.72 | 298.29 | 19 | 40 | 75.71 |
| SE | 21.16 | 214.51 | 7.53 | 11.97 | 35.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Segar, S.T.; Chantarasuwan, B.; Wong, D.-M.; Wang, R.; Chen, X.; Yu, H. Adaptation of Fig Wasps (Agaodinae) to Their Host Revealed by Large-Scale Transcriptomic Data. Insects 2021, 12, 815. https://doi.org/10.3390/insects12090815
Chen L, Segar ST, Chantarasuwan B, Wong D-M, Wang R, Chen X, Yu H. Adaptation of Fig Wasps (Agaodinae) to Their Host Revealed by Large-Scale Transcriptomic Data. Insects. 2021; 12(9):815. https://doi.org/10.3390/insects12090815
Chicago/Turabian StyleChen, Lianfu, Simon T. Segar, Bhanumas Chantarasuwan, Da-Mien Wong, Rong Wang, Xiaoyong Chen, and Hui Yu. 2021. "Adaptation of Fig Wasps (Agaodinae) to Their Host Revealed by Large-Scale Transcriptomic Data" Insects 12, no. 9: 815. https://doi.org/10.3390/insects12090815
APA StyleChen, L., Segar, S. T., Chantarasuwan, B., Wong, D.-M., Wang, R., Chen, X., & Yu, H. (2021). Adaptation of Fig Wasps (Agaodinae) to Their Host Revealed by Large-Scale Transcriptomic Data. Insects, 12(9), 815. https://doi.org/10.3390/insects12090815