
Comparative Mitogenomic Analysis of Five Awl Skippers (Lepidoptera: Hesperiidae: Coeliadinae) and Their Phylogenetic Implications



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequencing, Assembly, Annotation and Bioinformatic Analyses
2.3. Phylogenetic Analysis
3. Results and Discussion
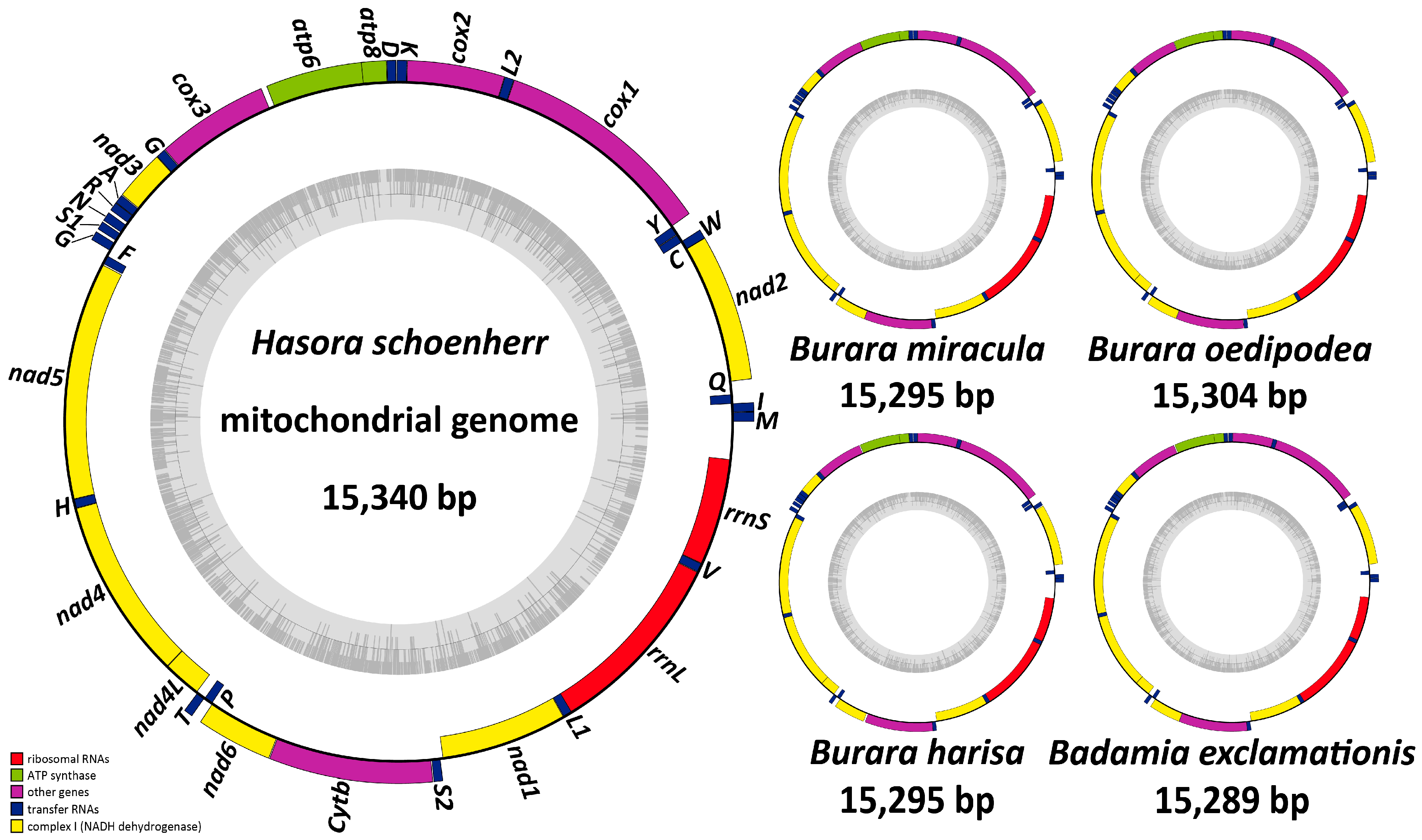
3.1. Structure and Nucleotide Composition of Mitogenome
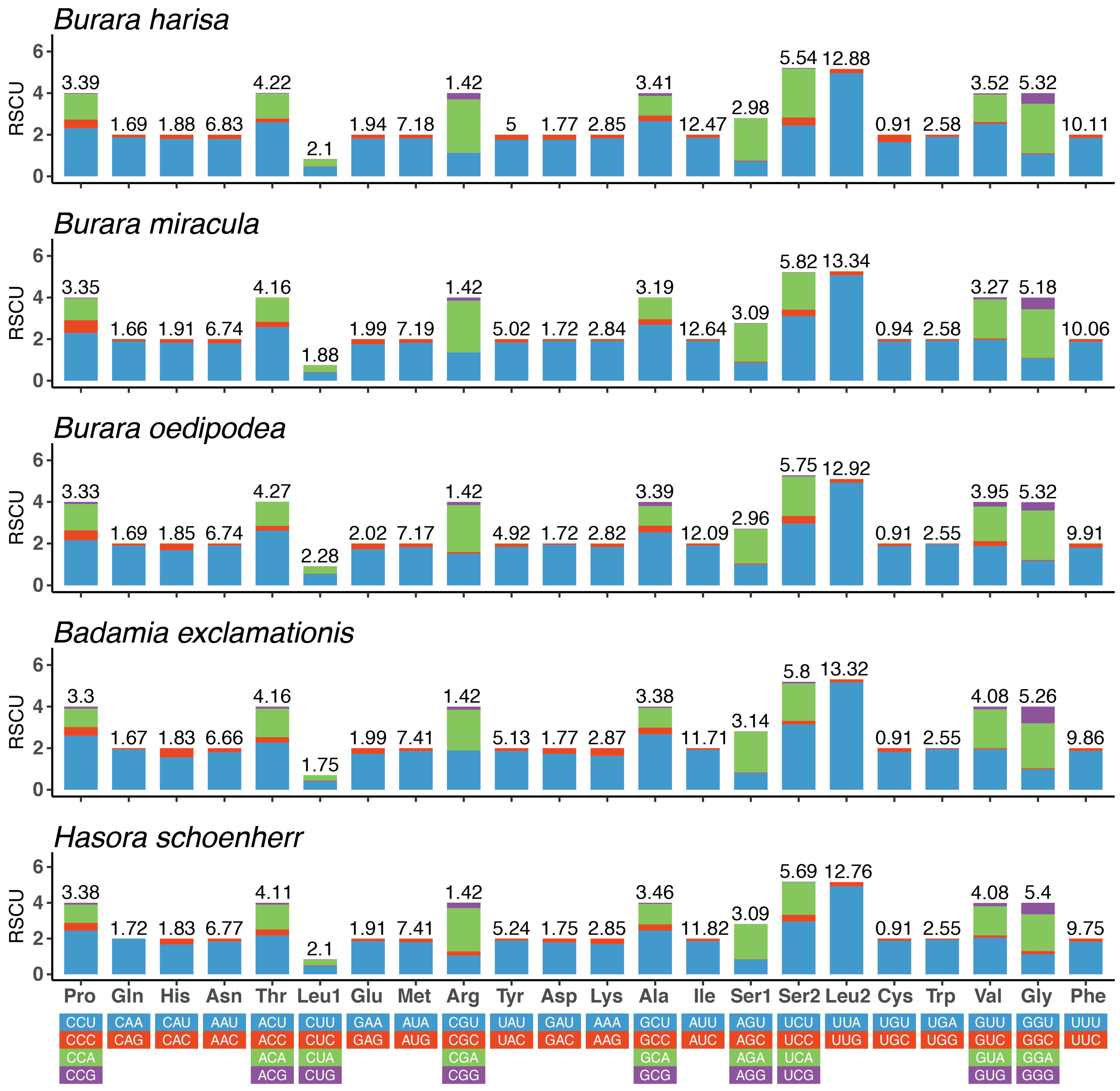
3.2. Protein-Coding Genes and Codon Usage
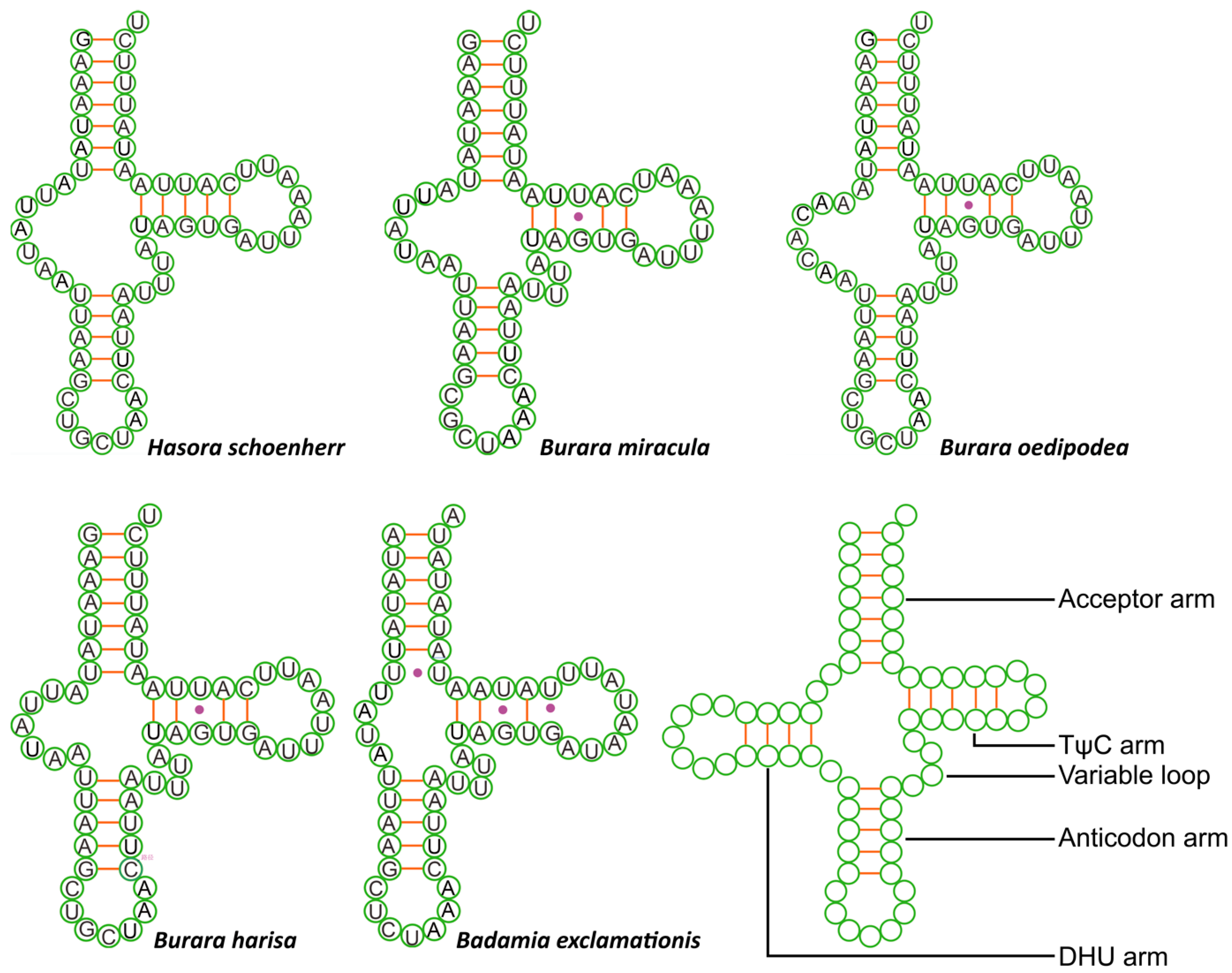
3.3. Transfer and Ribosomal RNA Genes
3.4. Overlapping Sequences, Intergenic Spacers and AT-Rich Region
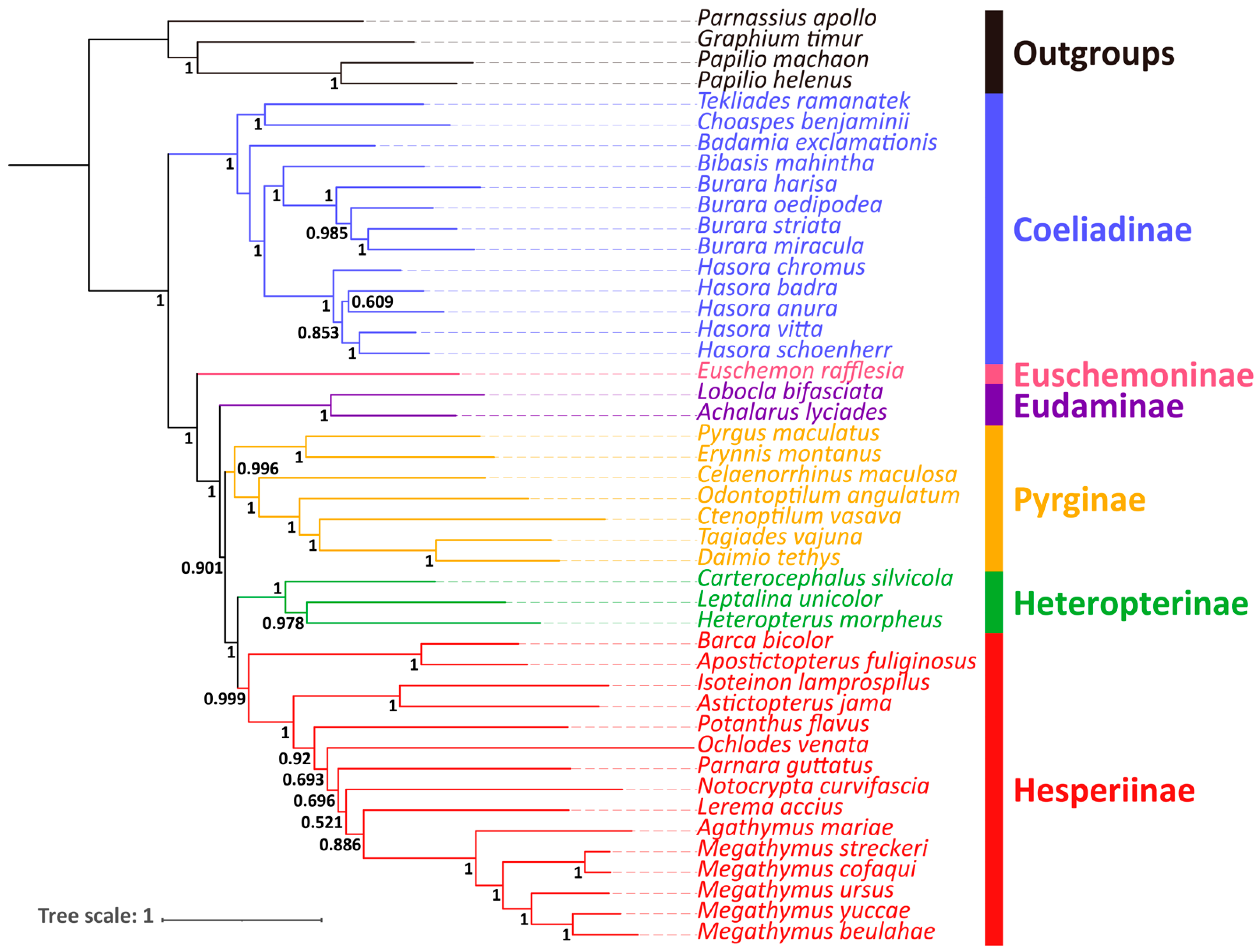
3.5. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G.D.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef]
- Zhang, D.; Rheindt, F.E.; She, H.; Cheng, Y.; Song, G.; Jia, C.; Qu, Y.; Alström, P.; Lei, F. Most genomic loci misrepresent the phylogeny of an avian radiation because of ancient gene flow. Syst. Biol. 2021, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Cong, Q.; Shen, J.; Brockmann, E.; Grishin, N.V. Three new subfamilies of skipper butterflies (Lepidoptera, Hesperiidae). Zookeys 2019, 861, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lees, D.C.; Shen, J.; Cong, Q.; Huertas, B.; Martin, G.; Grishin, N.V. The mitogenome of a Malagasy butterfly Malaza fastuosus (Mabille, 1884) recovered from the holotype collected over 140 years ago adds support for a new subfamily of Hesperiidae (Lepidoptera). Genome 2020, 63, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.; Liu, J.; Chiba, H.; Xiao, J.; Yuan, X. Complete mitochondrial genomes of three skippers in the tribe Aeromachini (Lepidoptera: Hesperiidae: Hesperiinae) and their phylogenetic implications. Ecol. Evol. 2021, 11, 8381–8393. [Google Scholar] [CrossRef]
- Chiba, H. A revision of the subfamily Coeliadinae (Lepidoptera: Hesperiidae). Bull. Kitakyushu Mus. Nat. Hist. Hum. Hist. Ser. A (Nat. Hist.) 2009, 7, 1–51. [Google Scholar]
- Warren, A.D.; Ogawa, J.R.; Brower, A.V.Z. Phylogenetic relationships of subfamilies and circumscription of tribes in the family Hesperiidae (Lepidoptera: Hesperioidea). Cladistics 2008, 24, 642–676. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, R.K.; Warren, A.D.; Collins, S.C.; Kodandaramaiah, U. Hostplant change and paleoclimatic events explain diversification shifts in skipper butterflies (Family: Hesperiidae). BMC Evol. Biol. 2017, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, E.F.A.; Breinholt, J.W.; Earl, C.; Warren, A.D.; Brower, A.V.Z.; Yago, M.; Dexter, K.M.; Espeland, M.; Pierce, N.E.; Lohman, D.J. Anchored phylogenomics illuminates the skipper butterfly tree of life. BMC Evol. Biol. 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cong, Q.; Shen, J.; Zhang, J.; Hallwachs, W.; Janzen, D.H.; Grishin, N.V. Genomes of skipper butterflies reveal extensive convergence of wing patterns. Proc. Natl. Acad. Sci. USA 2019, 116, 6232–6237. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Yuan, X.; Xue, G. Fauna Sinica: Insecta. Lepidoptera, Hesperiidae; Science Press: Beijing, China, 2015. [Google Scholar]
- Toussaint, E.F.A.; Chiba, H.; Yago, M.; Dexter, K.M.; Warren, A.D.; Storer, C.; Lohman, D.J.; Kawahara, A.Y. Afrotropics on the wing: Phylogenomics and historical biogeography of awl and policeman skippers. Syst. Entomol. 2021, 46, 172–185. [Google Scholar] [CrossRef]
- Drury, D. Illustrations of Natural History; White: London, UK, 1773; Volume 2. [Google Scholar]
- Zhang, J.; Cong, Q.; Fan, X.; Wang, R.; Wang, M.; Grishin, N.V. Mitogenomes of giant-skipper butterflies reveal an ancient split between deep and shallow root feeders. F1000Research 2017, 6, 222. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Wang, A.R.; Park, J.S.; Kim, I. Complete mitochondrial genomes of five skippers (Lepidoptera: Hesperiidae) and phylogenetic reconstruction of Lepidoptera. Gene 2014, 549, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; James John, Y.; Xuan, S.; Cao, T.; Yuan, X. The complete mitochondrial genome of the butterfly Hasora anura (Lepidoptera: Hesperiidae). Mitochondrial DNA Part A 2016, 27, 4401–4402. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, J.; James John, Y.; Yau Shiu, M.; Yuan, X.; Liu, J.; Cao, T. The complete mitochondrial genome of Hasora vitta (Butler, 1870) (Lepidoptera: Hesperiidae). Mitochondrial DNA Part A 2016, 27, 3020–3021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cong, Q.; Shen, J.; Fan, X.-L.; Wang, M.; Grishin, N.V. The complete mitogenome of Euschemon rafflesia (Lepidoptera: Hesperiidae). Mitochondrial DNA Part B 2017, 2, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Hao, J.; Zhao, H. Characterization of complete mitochondrial genome of the skipper butterfly, Celaenorrhinus maculosus (Lepidoptera: Hesperiidae). Mitochondrial DNA 2015, 26, 690–691. [Google Scholar] [CrossRef]
- Hao, J.; Sun, Q.; Zhao, H.; Sun, X.; Gai, Y.; Yang, Q. The complete mitochondrial genome of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae) and its phylogenetic implication. Comp. Funct. Genomics 2012, 2012, 328049. [Google Scholar] [CrossRef] [Green Version]
- Zuo, N.; Gan, S.; Chen, Y.; Hao, J. The complete mitochondrial genome of the Daimio tethys (Lepidoptera: Hesperoidea: Hesperiidae). Mitochondrial DNA Part A 2016, 27, 1099–1100. [Google Scholar] [CrossRef]
- Wang, A.R.; Jeong, H.C.; Han, Y.S.; Kim, I. The complete mitochondrial genome of the mountainous duskywing, Erynnis montanus (Lepidoptera: Hesperiidae): A new gene arrangement in Lepidoptera. Mitochondrial DNA 2014, 25, 93–94. [Google Scholar] [CrossRef]
- Liu, F.; Li, Y.; Jakovlić, I.; Yuan, X. Tandem duplication of two tRNA genes in the mitochondrial genome of Tagiades vajuna (Lepidoptera: Hesperiidae). Eur. J. Entomol. 2017, 114, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xiao, J.; Hao, X.; Yuan, X. Unique duplication of trnN in Odontoptilum angulatum (Lepidoptera: Pyrginae) and phylogeny within Hesperiidae. Insects 2021, 12, 348. [Google Scholar] [CrossRef]
- Shen, J.; Cong, Q.; Grishin, N.V. The complete mitogenome of Achalarus lyciades (Lepidoptera: Hesperiidae). Mitochondrial DNA Part B 2016, 1, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; Kim, M.J.; Jeong, N.R.; Kim, I. Complete mitochondrial genome of the silver stripped skipper, Leptalina unicolor (Lepidoptera: Hesperiidae). Mitochondrial DNA Part B 2019, 4, 3418–3420. [Google Scholar] [CrossRef]
- Han, Y.; Huang, Z.; Tang, J.; Chiba, H.; Fan, X. The complete mitochondrial genomes of two skipper genera (Lepidoptera: Hesperiidae) and their associated phylogenetic analysis. Sci. Rep. 2018, 8, 15762. [Google Scholar] [CrossRef] [Green Version]
- Cong, Q.; Grishin, N.V. The complete mitochondrial genome of Lerema accius and its phylogenetic implications. PeerJ 2016, 4, e1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Sun, Q.; Hao, J. The complete mitochondrial genome of Parara guttata (Lepidoptera: Hesperiidae). Mitochondrial DNA 2015, 26, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, F.; Chiba, H.; Yuan, X. The mitochondrial genomes of three skippers: Insights into the evolution of the family Hesperiidae (Lepidoptera). Genomics 2020, 112, 432–441. [Google Scholar] [CrossRef]
- Tang, M.; Tan, M.; Meng, G.; Yang, S.; Su, X.; Liu, S.; Song, W.; Li, Y.; Wu, Q.; Zhang, A.; et al. Multiplex sequencing of pooled mitochondrial genomes—a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 2014, 42, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Gan, S.; Shao, L.; Cheng, C.; Hao, J. The complete mitochondrial genome of the Pazala timur (Lepidoptera: Papilionidae: Papilioninae). Mitochondrial DNA Part A 2016, 27, 533–534. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, D.; Wang, Y.; Zhu, C.; Hao, J. The complete mitochondrial genome of the endangered Apollo butterfly, Parnassius apollo (Lepidoptera: Papilionidae) and its comparison to other Papilionidae species. J. Asia. Pac. Entomol. 2014, 17, 663–671. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication of animal mitochondrial DNA. Cell 1982, 28, 693–705. [Google Scholar] [CrossRef]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, D.A.; Iyer, V.N.; Moses, A.M.; Eisen, M.B. Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genet. 2006, 2, e173. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, R.K.; Warren, A.D.; Wahlberg, N.; Brower, A.V.Z.; Lukhtanov, V.A.; Kodandaramaiah, U. Ten genes and two topologies: An exploration of higher relationships in skipper butterflies (Hesperiidae). PeerJ 2016, 4, e2653. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.B.; Lockhart, P.J. Deciphering ancient rapid radiations. Trends Ecol. Evol. 2007, 22, 258–265. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Species | Accession Number/ DNA Voucher | References |
|---|---|---|---|
| Hesperiidae | |||
| Coeliadinae | Burara miracula | MZ502491 | This study |
| Burara oedipodea | MZ502492 | This study | |
| Burara harisa | MZ502490 | This study | |
| Burara striata | NC_034676 | [15] | |
| Badamia exclamationis | MZ502489 | This study | |
| Tekliades ramanatek | NVG-7871 | [4] | |
| Bibasis mahintha | NVG-7865 | [4] | |
| Choaspes benjaminii | NC_024647 | [26] | |
| Hasora chromus | NVG-17119G11 | [4] | |
| Hasora anura | KF881049 | [27] | |
| Hasora vitta | NC_027170 | [28] | |
| Hasora badra | NC_045249 | Unpublished | |
| Hasora schoenherr | MZ502493 | This study | |
| Euschemoninae | Euschemon rafflesia | NC_034231 | [29] |
| Pyrginae | Celaenorrhinus maculosus | NC_022853 | [30] |
| Ctenoptilum vasava | JF713818 | [31] | |
| Tagiades (=Daimio) Tethys | KJ813807 | [32] | |
| Erynnis montanus | NC_021427 | [33] | |
| Pyrgus maculatus | NC_030192 | Unpublished | |
| Tagiades vajuna | KX865091 | [34] | |
| Odontoptilum angulatum | MW381783 | [35] | |
| Eudaminae | Achalarus lyciades | NC_030602 | [36] |
| Lobocla bifasciata | KJ629166 | [26] | |
| Heteropterinae | Carterocephalus silvicola | NC_024646 | [26] |
| Heteropterus morpheus | NC_028506 | Unpublished | |
| Leptalina unicolour | MK265705 | [37] | |
| Hesperiinae | Apostictopterus fuliginosus | NC_039946 | [38] |
| Barca bicolor | NC_039947 | [38] | |
| Lerema accius | NC_029826 | [39] | |
| Ochlodes venata | HM243593 | Unpublished | |
| Parnara guttatus | NC_029136 | [40] | |
| Potanthus flavus | KJ629167 | [26] | |
| Astictopterus jama | MH763663 | [41] | |
| Isoteinon lamprospilus | MH763664 | [41] | |
| Notocrypta curvifascia | MH763665 | [41] | |
| Agathymus mariae | KY630504 | [15] | |
| Megathymus beulahae | KY630505 | [15] | |
| Megathymus cofaqui | KY630503 | [15] | |
| Megathymus streckeri | KY630501 | [15] | |
| Megathymus ursus | KY630502 | [15] | |
| Megathymus yuccae | KY630500 | [15] | |
| Outgroup | |||
| Papilionidae | Papilio machaon | NC_018047 | Unpublished |
| Papilio helenus | NC_025757 | [42] | |
| Graphium timur | NC_024098 | [43] | |
| Parnassius apollo | NC_024727 | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.; Yang, Y.; Hao, X.; Xiao, J.; Liu, J.; Yuan, X. Comparative Mitogenomic Analysis of Five Awl Skippers (Lepidoptera: Hesperiidae: Coeliadinae) and Their Phylogenetic Implications. Insects 2021, 12, 757. https://doi.org/10.3390/insects12080757
Sun Q, Yang Y, Hao X, Xiao J, Liu J, Yuan X. Comparative Mitogenomic Analysis of Five Awl Skippers (Lepidoptera: Hesperiidae: Coeliadinae) and Their Phylogenetic Implications. Insects. 2021; 12(8):757. https://doi.org/10.3390/insects12080757
Chicago/Turabian StyleSun, Qi, Yumeng Yang, Xiangyu Hao, Jintian Xiao, Jiaqi Liu, and Xiangqun Yuan. 2021. "Comparative Mitogenomic Analysis of Five Awl Skippers (Lepidoptera: Hesperiidae: Coeliadinae) and Their Phylogenetic Implications" Insects 12, no. 8: 757. https://doi.org/10.3390/insects12080757
APA StyleSun, Q., Yang, Y., Hao, X., Xiao, J., Liu, J., & Yuan, X. (2021). Comparative Mitogenomic Analysis of Five Awl Skippers (Lepidoptera: Hesperiidae: Coeliadinae) and Their Phylogenetic Implications. Insects, 12(8), 757. https://doi.org/10.3390/insects12080757