
The Primary Complete Mitochondrial Genome of the Lappet Moth Brahmophthalma hearseyi (Lepidoptera: Brahmaeidae) and Related Phylogenetic Analysis


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. PCR Amplification and Sequencing
2.4. Sequence Assembly, Annotation, and Analysis
2.5. Phylogenetic Analysis
3. Results
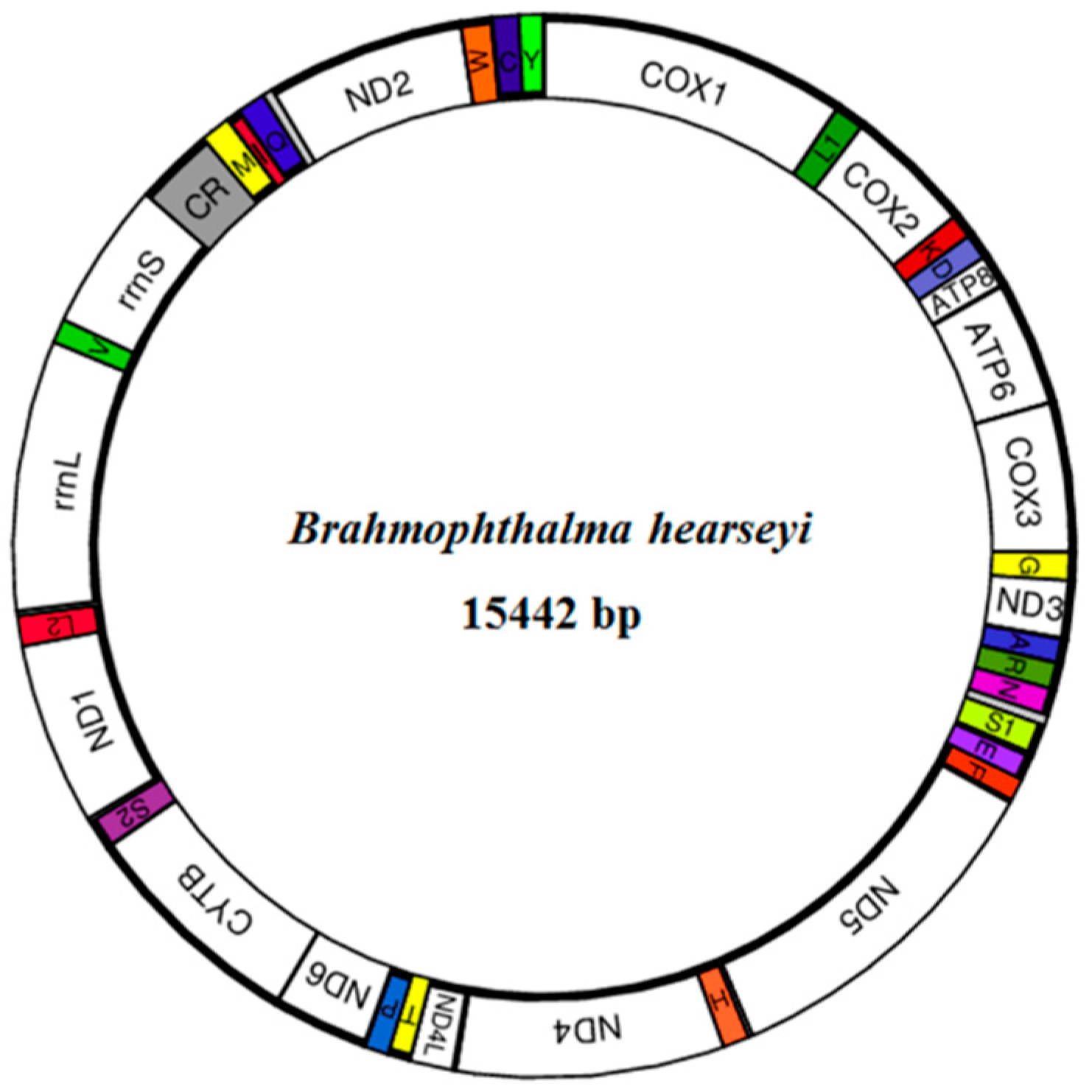
3.1. Genome Composition and Base Structure
3.2. PCGs
3.3. rRNA and tRNA Genes
3.4. A + T-Rich Region
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Graeber, M.B.; Müller, U. Recent developments in the molecular genetics of mitochondrial disorders. J. Neurol. Sci. 1998, 153, 251–263. [Google Scholar] [CrossRef]
- Liu, X.H.; Jian, X.U.; Zhou, J.; Xiao-Ping, L.U. The morphology of Brahmophthalma hearseyi white and the characters in Suzhou. J. Chang. Inst. Technol. 2009, 23, 69–70. [Google Scholar]
- Zhu, H.F.; Wang, L.Y. Chinese Brahmaeidae. Acta Entomol. Sin. 1977, 20, 83–84. [Google Scholar]
- Shimazaki, A.; Miyashita, T. Deer browsing reduces leaf damage by herbivorous insects through an induced response of the host plant. Ecol. Res. 2002, 17, 527–533. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef]
- Jiang, S.T.; Hong, G.Y.; Yu, M.; Li, N.; Yang, Y.; Liu, Y.Q.; Wei, Z.J. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int. J. Biol. Sci. 2009, 5, 351–365. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Yang, J. The complete mitochondrial genome of Rondotia menciana (Lepidoptera: Bombycidae). J. Insect Sci. 2015, 15, 48. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.Y.; Zhou, P.; Qiang, Y.; Qian, Z.Q. Characterization of the complete mitochondrial genome of Bombyx huttoni (Lepidoptera: Bombycidae). Mitochondrial DNA 2015, 27, 4112–4113. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, M.J.; Kim, I. The complete mitochondrial genome of the moon moth, Actias aliena (Lepidoptera: Saturniidae). Mitochondrial DNA Part A 2016, 27, 149–150. [Google Scholar] [CrossRef]
- Shantibala, T.; Victor, T.; Luikham, R.; Arunkumar, K.P.; Debaraj Sharma, H.; Lokeshwari, R.K.; Kim, I. Complete mitochondrial genome of the wild eri silkworm, Samia canningi (Lepidoptera: Saturniidae). Mitochondrial DNA Part A 2016, 27, 844–845. [Google Scholar] [CrossRef]
- Ackery, P.R. Systematic and faunistic studies on butterflies. In Symposia of the Royal Entomological Society of London; National Agricultural Library: Beltsville, MD, USA, 1984; pp. 304–306. [Google Scholar]
- Harvey, D.J. Higher Classification of the Nymphalidae, Appendix B. The Development and Evolution of Butterfly Wing Patterns; Nijhout, H.F., Ed.; Smithsonian Institution Press: Washington, DC, USA, 1991; pp. 255–273. [Google Scholar]
- Scoble, M.J. The Lepidoptera. Form, Function and Diversity; Oxford University Press: Oxford, UK, 1992; pp. 1–2. [Google Scholar]
- Heppner, J.B. Classification of Lepidoptera: Part 1: Introduction; Holarctic Lepidoptera; Association for Tropical Lepidoptera: Statesboro, GA, USA, 1998; p. 5. [Google Scholar]
- Wang, H.; Wahlberg, N.; Holloway, J.D.; Bergsten, J.; Fan, X.; Janzen, D.H.; Hallwachs, W.; Wen, L.; Wang, M.; Nylin, S. Molecular phylogeny of Lymantriinae (Lepidoptera, Noctuoidea, Erebidae) inferred from eight gene regions. Cladistics 2015, 31, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Mell, R. Beitrage Zur Fauna Sinica. V. Die Brahmaeiden und Eupterotiden Chinas; Deut. Ent. Zeit.: Berlin, Germany, 1929; pp. 337–487. [Google Scholar]
- Bryk, F. Brahmaeidae Hampson. Arkiv for Zoologi. Groszschmetterling Vom Korea 1949, 41, 1–125. [Google Scholar]
- Sauter, W. Zur systematis chen Stellung ron Brahmaea europaea Hartig (Lep. Brahmaeidae). Mitt. Schweiz. Ent. Ges. 1967, 40, 125–129. [Google Scholar]
- Hao, H.L.; Zhang, X.R.; Yang, J.K. Genus and Distribution of Brahmaeidae (Lepidoptera) from China. J. China Agric. Univ. 1999, 4, 37–42. [Google Scholar]
- Hao, J.; Li, C.; Sun, X.; Yang, Q. Phylogeny and divergence time estimation of cheilostome bryozoans based on mitochondrial 16S rRNA sequences. Chin. Sci. Bull. 2005, 50, 1205–1211. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Simmons, R.B.; Weller, S.J. Utility and evolution of cytochrome b in insects. Mol. Phylogenetics Evol. 2001, 20, 196–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Clewley, J.P. Macintosh sequence analysis software. Mol. Biotechnol. 1995, 3, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Yang, Z.; Rannala, B. Bayesian phylogenetic inference using DNA sequences: A Markov Chain Monte Carlo method. Mol. Biol. Evol. 1997, 14, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Tan, M.; Meng, G.; Yang, S.; Su, X.; Liu, S.; Zhou, X. Multiplex sequencing of pooled mitochondrial genomes—A crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 2014, 42, e166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Wu, A.Q. The complete mitochondrial genome of Bombyx mori strain Chunyun (Lepidoptera: Bombycidae). Mitochondrial DNA 2016, 27, 4082–4083. [Google Scholar] [CrossRef]
- Yukuhiro, K.; Sezutsu, H.; Itoh, M.; Shimizu, K.; Banno, Y. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol. Biol. Evol. 2002, 19, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Zhang, Y.; Zhou, X.; Kong, X.; Wei, S.; Ward, R.D.; Zhang, A.B. Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes. BMC Genom. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Song, L.; Shi, Y.; Yin, Y.; Wang, Y.; Zhang, P.; Chen, J.; Lou, L.; Liu, X. The complete mitochondrial genome of a medicinal insect, Hydrillodes repugnalis (Lepidoptera: Noctuoidea: Erebidae), and related phylogenetic analysis. Int. J. Biol. Macromol. 2019, 123, 485–493. [Google Scholar] [CrossRef]
- Liu, Q.N.; Zhu, B.J.; Dai, L.S.; Wei, G.Q.; Liu, C.L. The complete mitochondrial genome of the wild silkworm moth, Actias selene. Gene 2012, 505, 291–299. [Google Scholar] [CrossRef]
- Yang, L.; Wei, Z.J.; Hong, G.Y.; Jiang, S.T.; Wen, L.P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol. Biol. Rep. 2009, 36, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- MaKS, N. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Chen, M.M.; Li, Y.; Chen, M.; Wang, H.; Li, Q.; Xia, R.X.; Qin, L. Complete mitochondrial genome of the atlas moth, Attacus atlas (Lepidoptera: Saturniidae) and the phylogenetic relationship of Saturniidae species. Gene 2014, 545, 95–101. [Google Scholar] [CrossRef]
- Hong, G.; Jiang, S.; Yu, M.; Yang, Y.; Li, F.; Xue, F.; Wei, Z. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochim. Biophys. Sin. 2009, 41, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Kim, M.I.; Hong, M.Y.; Kim, K.Y.; Kang, P.D.; Hwang, J.S.; Kim, I. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae). Mol. Biol. Rep. 2009, 36, 1871–1880. [Google Scholar] [CrossRef]
- Lee, E.S.; Shin, K.S.; Kim, M.S.; Park, H.; Cho, S.; Kim, C.B. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 2006, 373, 52–57. [Google Scholar] [CrossRef]
- Sima, Y.H.; Chen, M.; Yao, R.; Li, Y.P.; Liu, T.; Jin, X.; Liu, Y.Q. The complete mitochondrial genome of the Ailanthus silkmoth, Samia cynthia cynthia (Lepidoptera: Saturniidae). Gene 2013, 526, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Pan, M.; Dai, F.; Zhu, X.; Lu, C.; Xiang, Z. The complete mitochondrial genome of the Chinese oak silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae). Acta Biochim. Biophys. Sin. 2008, 40, 693–703. [Google Scholar] [CrossRef]
- Coates, B.S.; Sumerford, D.V.; Hellmich, R.L. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnacalis. Int. J. Biol. Sci. 2005, 1, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.F.; McKechnie, S.W.; Pierce, N.; Kreitman, M. The lepidopteran mitochondrial control region: Structure and evolution. Mol. Biol. Evol. 1993, 10, 1259–1272. [Google Scholar] [PubMed] [Green Version]
- Liu, Y.; Xin, Z.Z.; Zhu, X.Y.; Zhao, X.M.; Wang, Y.; Tang, B.P.; Zhang, H.B.; Zhang, D.Z.; Zhou, C.L.; Liu, Q.N. The complete mitochondrial genome of Euproctis similis (Lepidoptera: Noctuoidea: Erebidae) and phylogenetic analysis. Int. J. Biol. Macromol. 2017, 105 Pt 1, 219–227. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Lin, R.; Zhang, Y.; Hu, K.; Li, Y.; Huang, Z.; Peng, S.; Ding, J.H.; Geng, X.X.; et al. Mitochondrial genome characteristics of Somena scintillans (Lepidoptera: Erebidae) and comparation with other Noctuoidea insects. Genomics 2019, 111, 1239–1248. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect Mitochondrial Control rengion: A Review of structure, Evolitionar ystudies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Wu, Q.L.; Gong, Y.J.; Gu, Y.; Wei, S.J. The complete mitochondrial genome of the beet armyworm Spodoptera exigua (Hübner) (Lepodiptera: Noctuidae). Mitochondrial DNA 2013, 24, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef]
- Tillyard, R.J.; Dunstan, B. Mesozoic insects of Queensland; Government Printer: Cumming, AJ, USA, 1924; Volume 2.
- Kuznetzov, V.I.; Stekolnikov, A.A. Comparative and functional morphology of the male genitalia of the bombycoid moths (Lepidoptera, Papilionomorpha: Lasiocampoidea, Sphingoidea, Bombycoidea) and their systematic position. Tr. Zool. Inst. Leningr. 1985, 134, 3–48. [Google Scholar]
- Minet, J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata). Insect Syst. Evol. 1991, 22, 69–95. [Google Scholar] [CrossRef]
- Brock, J.P. A contribution towards an understanding of the morphology and phylogeny of the Ditrysian Lepidoptera. J. Nat. Hist. 1971, 5, 29–102. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Family | Species | Whole Genome | PCG | rRNA | GenBank Accession No. | References | |
|---|---|---|---|---|---|---|---|
| Size | A + T | A + T | A + T | ||||
| (bp) | (%) | (%) | (%) | ||||
| Saturniidae | Actias selene | 15,236 | 78.91 | 77.36 | 83.73 | NC_018133 | Liu 2012 |
| Saturniidae | Antheraea pernyi | 15,566 | 80.16 | 78.47 | 83.87 | NC_004622 | Liu 2008 |
| Saturniidae | Antheraea yamamai | 15,338 | 80.29 | 78.94 | 84.14 | EU726630 | Kim 2009 |
| Geometridae | Biston panterinaria | 15,517 | 79.55 | 77.39 | 85.27 | NC_020004 | Yang 2012 |
| Geometridae | Biston suppressaria | 15,628 | 79.43 | 77.28 | 84.92 | NC_027111 | Chen 2015 |
| Bombycidae | Bombyx mandarina | 15,928 | 81.68 | 79.69 | 85.19 | NC_003395 | Yukuhiro 2002 |
| Bombycidae | Bombyx mori | 15,666 | 81.35 | 79.57 | 84.82 | KM875545 | Zhang 2014 |
| Brahmaeidae | Brahmophthalma hearseyi | 15,442 | 80.81 | 79.27 | 83.87 | KU884326 | This study |
| Lasiocampidae | Dendrolimus punctatus | 15,411 | 79.46 | 77.62 | 84.73 | NC_027156 | Qin 2015 |
| Lasiocampidae | Dendrolimus spectabilis | 15,411 | 79.5 | 77.71 | 83.68 | NC_025763 | Tang 2011 |
| Pyralidae | Ephestia kuehniella | 15,295 | 79.76 | 78.18 | 84.44 | NC_022476 | Traut 2013 |
| Saturniidae | Eriogyna pyretorum | 15,327 | 80.82 | 79.41 | 84.55 | NC_012727 | Jiang 2009 |
| Lymantriidae | Euproctis pseudoconspersa | 15,461 | 79.93 | 77.99 | 84.87 | NC_027145 | Dong 2016 |
| Pyralidae | Galleria mellonella | 15,320 | 80.42 | 78.88 | 84.37 | NC_028532 | unpublished |
| Crambidae | Haritalodes derogata | 15,235 | 80.7 | 79.19 | 84.59 | NC_029202 | Zhao 2015 |
| Geometridae | Jankowskia athleta | 15,534 | 79.53 | 77.71 | 83.83 | NC_027948 | Xu 2015 |
| Lymantriidae | Lymantria dispar | 15,569 | 79.88 | 77.84 | 84.6 | NC_012893 | Zhu 2010 |
| Sphingidae | Manduca sexta | 15,516 | 81.79 | 80.3 | 85.42 | NC_010266 | Cameron 2008 |
| Geometridae | Phthonandria atrilineata | 15,499 | 81.02 | 79.1 | 85.93 | NC_010522 | Yang 2009 |
| Bombycidae | Rondotia menciana | 15,301 | 78.86 | 77.1 | 83.74 | NC_021962 | Kong 2015 |
| Sphingidae | Sphinx morio | 15,299 | 81.17 | 79.84 | 84.8 | NC_020780 | Min 2013 |
| Noctuidae | Spodoptera exigua | 15,365 | 80.93 | 79.47 | 85.15 | NC_019622 | Qiu-Ling 2013 |
| Noctuidae | Spodoptera litura | 15,388 | 80.98 | 79.55 | 84.71 | NC_022676 | Wan 2013 |
| Crambidae | Spoladea recurvalis | 15,273 | 80.89 | 79.37 | 85.54 | NC_027443 | He 2014 |
| Drosophilidae | Drosophila melanogaster | 19,517 | 82.16 | 77.23 | 81.9 | U37541 | Clary 1982 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Gao, S.; Cai, S.; Zou, Z.; Xin, T.; Xia, B. The Primary Complete Mitochondrial Genome of the Lappet Moth Brahmophthalma hearseyi (Lepidoptera: Brahmaeidae) and Related Phylogenetic Analysis. Insects 2021, 12, 973. https://doi.org/10.3390/insects12110973
Yang S, Gao S, Cai S, Zou Z, Xin T, Xia B. The Primary Complete Mitochondrial Genome of the Lappet Moth Brahmophthalma hearseyi (Lepidoptera: Brahmaeidae) and Related Phylogenetic Analysis. Insects. 2021; 12(11):973. https://doi.org/10.3390/insects12110973
Chicago/Turabian StyleYang, Shan, Shangren Gao, Shiyu Cai, Zhiwen Zou, Tianrong Xin, and Bin Xia. 2021. "The Primary Complete Mitochondrial Genome of the Lappet Moth Brahmophthalma hearseyi (Lepidoptera: Brahmaeidae) and Related Phylogenetic Analysis" Insects 12, no. 11: 973. https://doi.org/10.3390/insects12110973
APA StyleYang, S., Gao, S., Cai, S., Zou, Z., Xin, T., & Xia, B. (2021). The Primary Complete Mitochondrial Genome of the Lappet Moth Brahmophthalma hearseyi (Lepidoptera: Brahmaeidae) and Related Phylogenetic Analysis. Insects, 12(11), 973. https://doi.org/10.3390/insects12110973