Transcriptomic Analysis of Mating Responses in Bemisia tabaci MED Females




 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Rearing and Sample Preparation
2.2. RNA Extraction, cDNA Library Construction, and Illumina Sequencing
2.3. Comparative Analysis and Functional Annotation
2.4. Identification of Mating-Related DEGs and Enrichment Analysis
2.5. RT-qPCR Analysis
2.6. Mating-Related Genes Expression Profiles Analysis at Different Time Points
2.7. Statistical Analysis
3. Results
3.1. Illumina Sequencing and Clean-Read Map
3.2. Functional Annotation and Classification
3.3. Statistics Analysis of Mating-Related DEGs
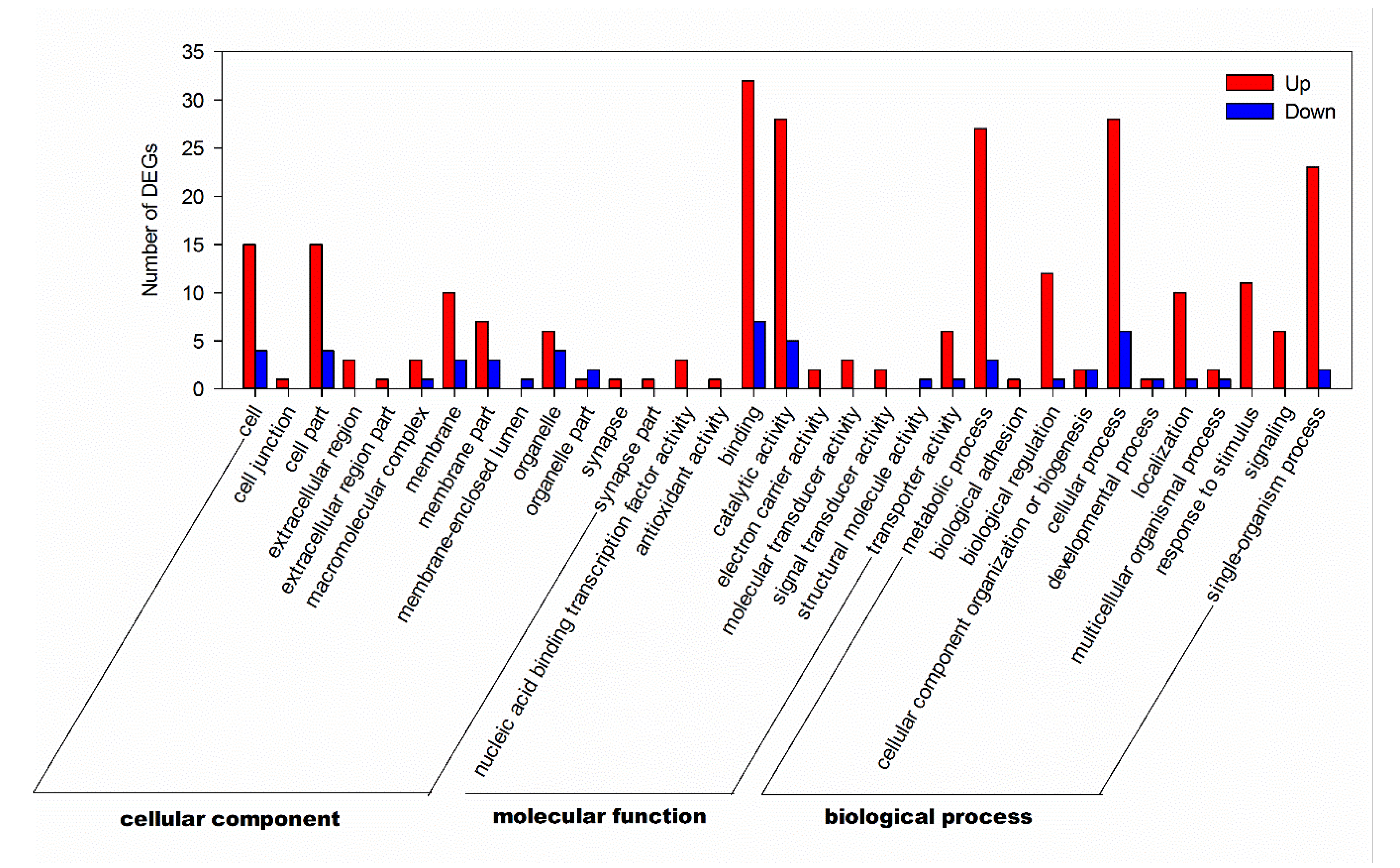
3.4. GO Annotation and KEGG Pathways Analysis of DEGs
3.5. Real-Time PCR Validation of Mating-Related Genes
3.6. Expression Profiles of Mating Related Genes at Different Time Points
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lawniczak, M.K.; Begun, D.J. A genome-wide analysis of courting and mating responses in Drosophila melanogaster females. Genome 2004, 47, 900–910. [Google Scholar] [CrossRef][Green Version]
- Kocher, S.D.; Richard, F.J.; Tarpy, D.R.; Grozinger, C.M. Genomic analysis of post-mating changes in the honey bee queen (Apis mellifera). BMC Genom. 2008, 9, 232. [Google Scholar] [CrossRef]
- Rogers, D.W.; Whitten, M.M.; Thailayil, J.; Soichot, J.; Levashina, E.A.; Catteruccia, F. Molecular and cellular components of the mating machinery in Anopheles gambiae females. Proc. Natl. Acad. Sci. USA 2008, 105, 19390–19395. [Google Scholar] [CrossRef]
- Innocenti, P.; Morrow, E.H. Immunogenic males: A genome-wide analysis of reproduction and the cost of mating in Drosophila melanogaster females. J. Evol. Biol. 2009, 22, 964–973. [Google Scholar] [CrossRef]
- Gomulski, L.M.; Dimopoulos, G.; Xi, Z.; Scolari, F.; Gabrieli, P.; Siciliano, P.; Clarke, A.R.; Malacrida, A.R.; Gasperi, G. Transcriptome profiling of sexual maturation and mating in the Mediterranean fruit fly, Ceratitis capitata. PLoS ONE 2012, 7, e30857. [Google Scholar] [CrossRef]
- Short, S.M.; Lazzaro, B.P. Reproductive status alters transcriptomic response to infection in female Drosophila melanogaster. G3 Genes Genom. Genet. 2013, 3, 827–840. [Google Scholar]
- Alfonso-Parra, C.; Ahmed-Braimah, Y.H.; Degner, E.C.; Avila, F.W.; Villarreal, S.M.; Pleiss, J.A.; Wolfner, M.F.; Harrington, M.F.; Harrington, L.C. Mating-induced transcriptome changes in the reproductive tract of female Aedes aegypti. PLoS Neglect. Trop. Dis. 2016, 10, e0004451. [Google Scholar] [CrossRef]
- Kapelnikov, A.; Zelinger, E.; Gottlieb, Y.; Rhrissorrakrai, K.; Gunsalus, K.C.; Heifetz, Y. Mating induces an immune response and developmental switch in the Drosophila oviduct. Proc. Natl. Acad. Sci. USA 2008, 105, 13912–13917. [Google Scholar] [CrossRef]
- Zelazny, B. Transmission of a baculovirus in populations of Oryctes rhinoceros. J. Invertebr. Pathol. 1976, 27, 221–227. [Google Scholar] [CrossRef]
- Fuchs, M.S.; Craig, G.B., Jr.; Hiss, E.A. The biochemical basis of female monogamy in mosquitoes: I. Extraction of the active principle from Aedes aegypti. Life Sci. 1968, 7, 835–839. [Google Scholar] [CrossRef]
- Judson, C.L. Feeding and oviposition behavior in the mosquito Aedes aegypti (L.). I. Preliminary studies of physiological control mechanisms. Biol. Bull. 1967, 133, 369–377. [Google Scholar] [CrossRef]
- Helinski, M.E.; Harrington, L.C. Male mating history and body size influence female fecundity and longevity of the dengue vector Aedes aegypti. J. Med. Entomol. 2011, 48, 202–211. [Google Scholar] [CrossRef]
- Thailayil, J.; Magnusson, K.; Godfray, H.C.J.; Crisanti, A.; Catteruccia, F. Spermless males elicit large-scale female responses to mating in the malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2011, 108, 13677–13681. [Google Scholar] [CrossRef]
- Gabrieli, P.; Kakani, E.G.; Mitchell, S.N.; Mameli, E.; Want, E.J.; Anton, A.M.; Serrao, A.; Baldini, F.; Catteruccia, F. Sexual transfer of the steroid hormone 20E induces the postmating switch in Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2014, 111, 16353–16358. [Google Scholar] [CrossRef]
- De Barro, P.J.; Liu, S.S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef]
- Gilbertson, R.L.; Batuman, O.; Webster, C.G.; Adkins, S. Role of the insect supervectors Bemisia tabaci and Frankliniella occidentalis in the emergence and global spread of plant viruses. Annu. Rev. Virol. 2015, 2, 67–93. [Google Scholar] [CrossRef]
- Luan, J.B.; Ruan, Y.M.; Zhang, L.; Liu, S.S. Pre-copulation intervals, copulation frequencies, and initial progeny sex ratios in two biotypes of whitefly, Bemisia tabaci. Entomol. Exp. Appl. 2008, 129, 316–324. [Google Scholar] [CrossRef]
- Liu, S.S.; De Barro, P.J.; Xu, J.; Luan, J.B.; Zang, L.S.; Ruan, Y.M.; Wan, F.H. Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science 2007, 318, 1769–1772. [Google Scholar] [CrossRef]
- Chu, D.; Zhang, Y.J.; Cong, B.; Xu, B.Y.; Wu, Q.J.; Zhu, G.R. Sequence analysis of mtDNA COI gene and molecular phylogeny of different geographical populations of Bemisia tabaci (Gennadius). Agr. Sci. China 2005, 4, 533–541. [Google Scholar]
- Xie, W.; Chen, C.; Yang, Z.; Guo, L.; Yang, X.; Wang, D.; Chen, M.; Huang, J.; Wen, Y.; Zeng, Y.; et al. Genome sequencing of the sweetpotato whitefly Bemisia tabaci MED/Q. GigaScience 2017, 6, gix018. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Li, R.; Xie, W.; Wang, S.; Wu, Q.; Yang, N.; Yang, X.; Pan, H.; Zhou, X.; Bai, L.; Xu, B.; et al. Reference gene selection for qRT-PCR analysis in the sweetpotato whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae). PLoS ONE 2013, 8, e53006. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mack, P.D.; Kapelnikov, A.; Heifetz, Y.; Bender, M. Mating-responsive genes in reproductive tissues of female Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2006, 103, 10358–10363. [Google Scholar] [CrossRef]
- Kocher, S.D.; Tarpy, D.R.; Grozinger, C.M. The effects of mating and instrumental insemination on queen honey bee flight behaviour and gene expression. Insect Mol. Biol. 2010, 19, 153–162. [Google Scholar] [CrossRef]
- Blackman, R.; Cahill, M. The karyotype of Bemisia tabaci (Hemiptera: Aleyrodidae). Bull. Entomol. Res. 1998, 88, 213–215. [Google Scholar] [CrossRef]
- Anto, P.R.; Per, Q.; Ross, L.; Francesco, L.; Jia, J.; Mette, N.; Ole, M.; Anders, D.B.; Qibin, L.; Jane, H.C. Experimental validation of methods for differential gene expression analysis and sample pooling in RNA-seq. Bmc Genom. 2015, 16, 548. [Google Scholar]
- McGraw, L.A.; Gibson, G.; Clark, A.G.; Wolfner, M.F. Genes regulated by mating, sperm, or seminal proteins in mated female Drosophila melanogaster. Curr. Biol. 2004, 14, 1509–1514. [Google Scholar] [CrossRef]
- Nie, H.; Jiang, K.; Jiang, L.; Huo, Z.; Ding, J.; Yan, X. Transcriptome analysis reveals the pigmentation related genes in four different shell color strains of the Manila clam Ruditapes philippinarum. Genomics 2020, 112, 2011–2020. [Google Scholar] [CrossRef]
- Feyereisen, R. Insect P450 enzymes. Annu. Rev. Entomol. 1999, 44, 507–533. [Google Scholar] [CrossRef]
- Chapman, T.; Liddle, L.F.; Kalb, J.M.; Wolfner, M.F.; Partridge, L. Cost of mating in Drosophila melanogaster females is mediated by male accessory gland products. Nature 1995, 373, 241–244. [Google Scholar] [CrossRef]
- Lung, O.; Kuo, L.; Wolfner, M.F. Drosophila males transfer antibacterial proteins from their accessory gland and ejaculatory duct to their mates. J. Insect Physiol. 2001, 47, 617–622. [Google Scholar] [CrossRef]
- Altincicek, B.; Knorr, E.; Vilcinskas, A. Beetle immunity: Identification of immune-inducible genes from the model insect Tribolium castaneum. Dev. Comp. Immunol. 2008, 32, 585–595. [Google Scholar] [CrossRef]
- Guittard, E.; Blais, C.; Maria, A.; Parvy, J.P.; Pasricha, S.; Lumb, C.; Lafont, R.; Daborn, P.J.; Dauphin-Villemant, C. CYP18A1, a key enzyme of Drosophila steroid hormone inactivation, is essential for metamorphosis. Dev. Biol. 2011, 349, 35–45. [Google Scholar] [CrossRef]
- Kristensen, T.N.; Dahlgaard, J.; Loeschcke, V. Inbreeding affects Hsp70 expression in two species of Drosophila even at benign temperatures. Evol. Ecol. Res. 2002, 4, 1209–1216. [Google Scholar]
- King, A.M.; MacRae, T.H. Insect heat shock proteins during stress and diapause. Annu. Rev. Entomol. 2015, 60, 59–75. [Google Scholar] [CrossRef]
- Boilard, M.; Reyes-Moreno, C.; Lachance, C.; Massicotte, L.; Bailey, J.L.; Sirard, M.A.; Leclerc, P. Localization of the chaperone proteins GRP78 and HSP60 on the luminal surface of bovine oviduct epithelial cells and their association with spermatozoa. Biol. Reprod. 2004, 71, 1879–1889. [Google Scholar] [CrossRef]
- Himuro, C.; Fujisaki, K. Mating experience weakens starvation tolerance in the seed bug Togo hemipterus (Heteroptera: Lygaeidae). Physiol. Entomol. 2010, 35, 128–133. [Google Scholar] [CrossRef]
- Wexler, Y.; Wertheimer, K.O.; Subach, A.; Pruitt, J.N.; Scharf, I. Mating alters the link between movement activity and pattern in the red flour beetle. Physiol. Entomol. 2017, 42, 299–306. [Google Scholar] [CrossRef]
- Shukla, E.; Thorat, L.J.; Nath, B.B.; Gaikwad, S.M. Insect trehalase: Physiological significance and potential applications. Glycobiology 2014, 25, 357–367. [Google Scholar] [CrossRef]
- Kikawada, T.; Saito, A.; Kanamori, Y.; Nakahara, Y.; Iwata, K.I.; Tanaka, D.; Watanabe, M.; Okuda, T. Trehalose transporter 1, a facilitated and high-capacity trehalose transporter, allows exogenous trehalose uptake into cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11585–11590. [Google Scholar] [CrossRef]
- Kanamori, Y.; Saito, A.; Hagiwara-Komoda, Y.; Tanaka, D.; Mitsumasu, K.; Kikuta, S.; Watanabe, M.; Cornette, R.; Kikawada, T.; Okuda, T. The trehalose transporter 1 gene sequence is conserved in insects and encodes proteins with different kinetic properties involved in trehalose import into peripheral tissues. Insect Biochem. Mol. Biol. 2010, 40, 30–37. [Google Scholar] [CrossRef]
- Thomas, A.W.; Gooding, R.H. Digestive Processes Of Hematophagous Insects: VIII. Estimation of meal size and demonstration of trypsin in horse flies and deer flies (Diptera: Tabanidae). J. Med. Entomol. 1976, 13, 131–136. [Google Scholar] [CrossRef]
- Buhi, W.C.; Alvarez, I.M.; Kouba, A.J. Secreted proteins of the oviduct. Cells Tissues Organs 2000, 166, 165–179. [Google Scholar] [CrossRef]
- Mayer, J.; Layfield, R.; Ardley, H.C.; Robinson, P.A. E3 ubiquitin ligases. Essays Biochem. 2005, 41, 15–30. [Google Scholar] [CrossRef]
- Ferveur, J.F. Cuticular hydrocarbons: Their evolution and roles in Drosophila pheromonal communication. Behav. Genet. 2005, 35, 279–295. [Google Scholar] [CrossRef]
- Wicker-Thomas, C. Pheromonal communication involved in courtship behavior in Diptera. J. Insect Physiol. 2007, 53, 1089–1100. [Google Scholar] [CrossRef]
- Yew, J.Y.; Chung, H. Insect pheromones: An overview of function, form, and discovery. Prog. Lipid Res. 2015, 59, 88–105. [Google Scholar] [CrossRef]
- Tsfadia, O.; Azrielli, A.; Falach, L.; Zada, A.; Roelofs, W.; Rafaeli, A. Pheromone biosynthetic pathways: PBAN-regulated rate-limiting steps and differential expression of desaturase genes in moth species. Insect Biochem. Mol. Biol. 2008, 38, 552–567. [Google Scholar] [CrossRef]
- Cha, W.H.; Jung, C.R.; Hwang, Y.J.; Lee, D.W. Comparative transcriptome analysis of pheromone biosynthesis-related gene expressions in Plutella xylostella (L.). J. Asia Pac. Entomol. 2017, 20, 1260–1266. [Google Scholar] [CrossRef]
- Liu, Q.N.; Zhu, B.J.; Wang, L.; Wei, G.Q.; Dai, L.S.; Lin, K.Z.; Sun, Y.; Qiu, J.F.; Fu, W.W.; Liu, C.L. Identification of immune response-related genes in the Chinese oak silkworm, Antheraea pernyi by suppression subtractive hybridization. J. Invertebr. Pathol. 2013, 114, 313–323. [Google Scholar] [CrossRef]
- Davis, S.; Anak, D.; Parish, C.R. Heparan sulfate: A ubiquitous glycosaminoglycan with multiple roles in immunity. Front. Immunol. 2013, 4, 470. [Google Scholar]
- Wang, Z.; Wu, Z.; Jian, J.; Lu, Y. Cloning and expression of heat shock protein 70 gene in the haemocytes of pearl oyster (Pinctada fucata, Gould 1850) responding to bacterial challenge. Fish Shellfish Immunol. 2009, 26, 639–645. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, D.; Luo, P.; He, X. Identification of the immune expressed sequence tags of pearl oyster (Pinctada martensii, Dunker 1850) responding to Vibrio alginolyticus challenge by suppression subtractive hybridization. Comp. Biochem. Physiol. D Genom. Proteom. 2012, 7, 243–247. [Google Scholar] [CrossRef]
- Aggarwal, K.; Silverman, N. Positive and negative regulation of the Drosophila immune response. BMB Rep. 2008, 41, 267–277. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Databases | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| DEGs | 434 | - | - | - | - | - | - | - | - |
| DEGs-annotated | 372 | 111 | 77 | 163 | 206 | 372 | 255 | 218 | 307 |
| Upregulation | 331 | - | 66 | 68 | - | - | - | - | - |
| Downregulation | 103 | - | 11 | 15 | - | - | - | - | - |
| KEGG Pathway | koID | DEGs Numbers | p-Value | Corrected p-Value |
|---|---|---|---|---|
| Longevity regulating pathway—Multiple species | ko04213 | 7 | 0.0005238 | 0.0413826 |
| Protein processing in endoplasmic reticulum | ko04141 | 10 | 0.0025248 | 0.1994616 |
| Pyruvate metabolism | ko00620 | 5 | 0.0043336 | 0.3423515 |
| Phosphatidylinositol signaling system | ko04070 | 5 | 0.0125282 | 0.9897258 |
| Endocytosis | ko04144 | 8 | 0.0218859 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, Z.; Liu, Y.; Yang, J.; Xie, W.; Wang, S.; Wu, Q.; Zhou, X.; Pang, B.; Zhang, Y. Transcriptomic Analysis of Mating Responses in Bemisia tabaci MED Females. Insects 2020, 11, 308. https://doi.org/10.3390/insects11050308
Huo Z, Liu Y, Yang J, Xie W, Wang S, Wu Q, Zhou X, Pang B, Zhang Y. Transcriptomic Analysis of Mating Responses in Bemisia tabaci MED Females. Insects. 2020; 11(5):308. https://doi.org/10.3390/insects11050308
Chicago/Turabian StyleHuo, Zhijia, Yating Liu, Jinjian Yang, Wen Xie, Shaoli Wang, Qingjun Wu, Xuguo Zhou, Baoping Pang, and Youjun Zhang. 2020. "Transcriptomic Analysis of Mating Responses in Bemisia tabaci MED Females" Insects 11, no. 5: 308. https://doi.org/10.3390/insects11050308
APA StyleHuo, Z., Liu, Y., Yang, J., Xie, W., Wang, S., Wu, Q., Zhou, X., Pang, B., & Zhang, Y. (2020). Transcriptomic Analysis of Mating Responses in Bemisia tabaci MED Females. Insects, 11(5), 308. https://doi.org/10.3390/insects11050308