Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis



 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Harmonia Axyridis Collection and DNA Extraction
2.2. Library Preparation and SNP Identification
2.3. Population Genetic Analyses
2.4. Construction of Simulated Populations
2.5. Optimizing Sample Size
3. Results
3.1. Population Characterization and SNP Identification
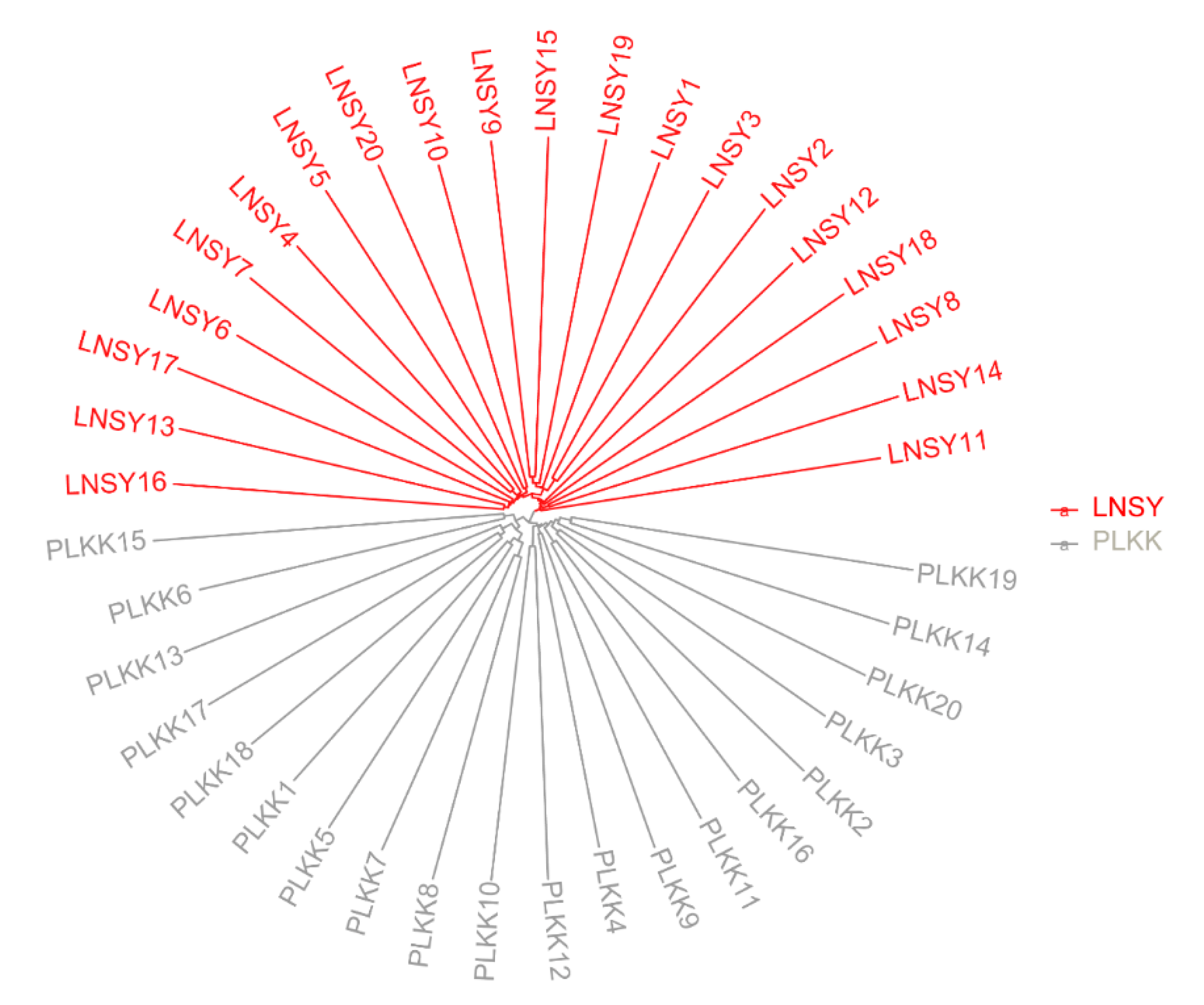
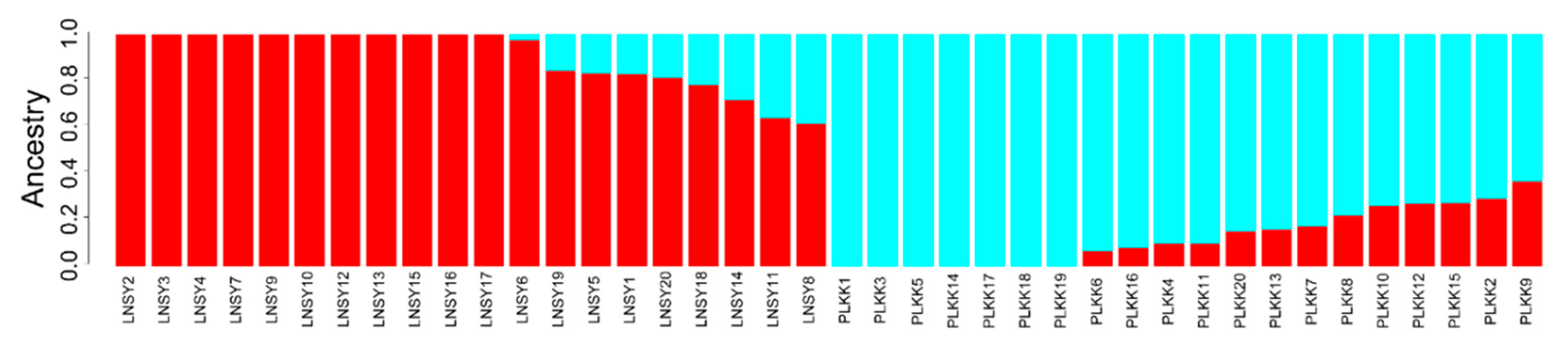
3.2. Population Genetic Structure
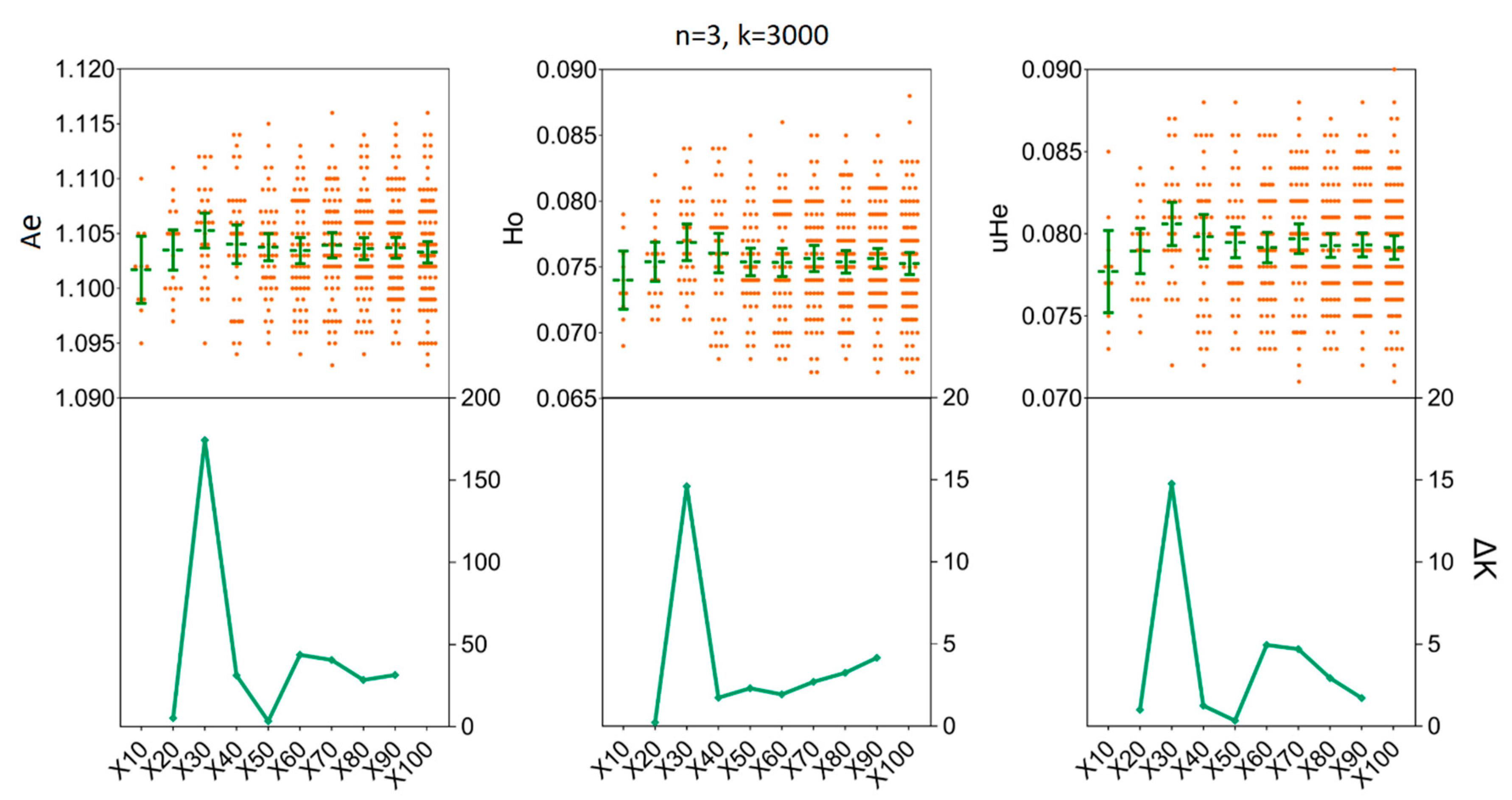
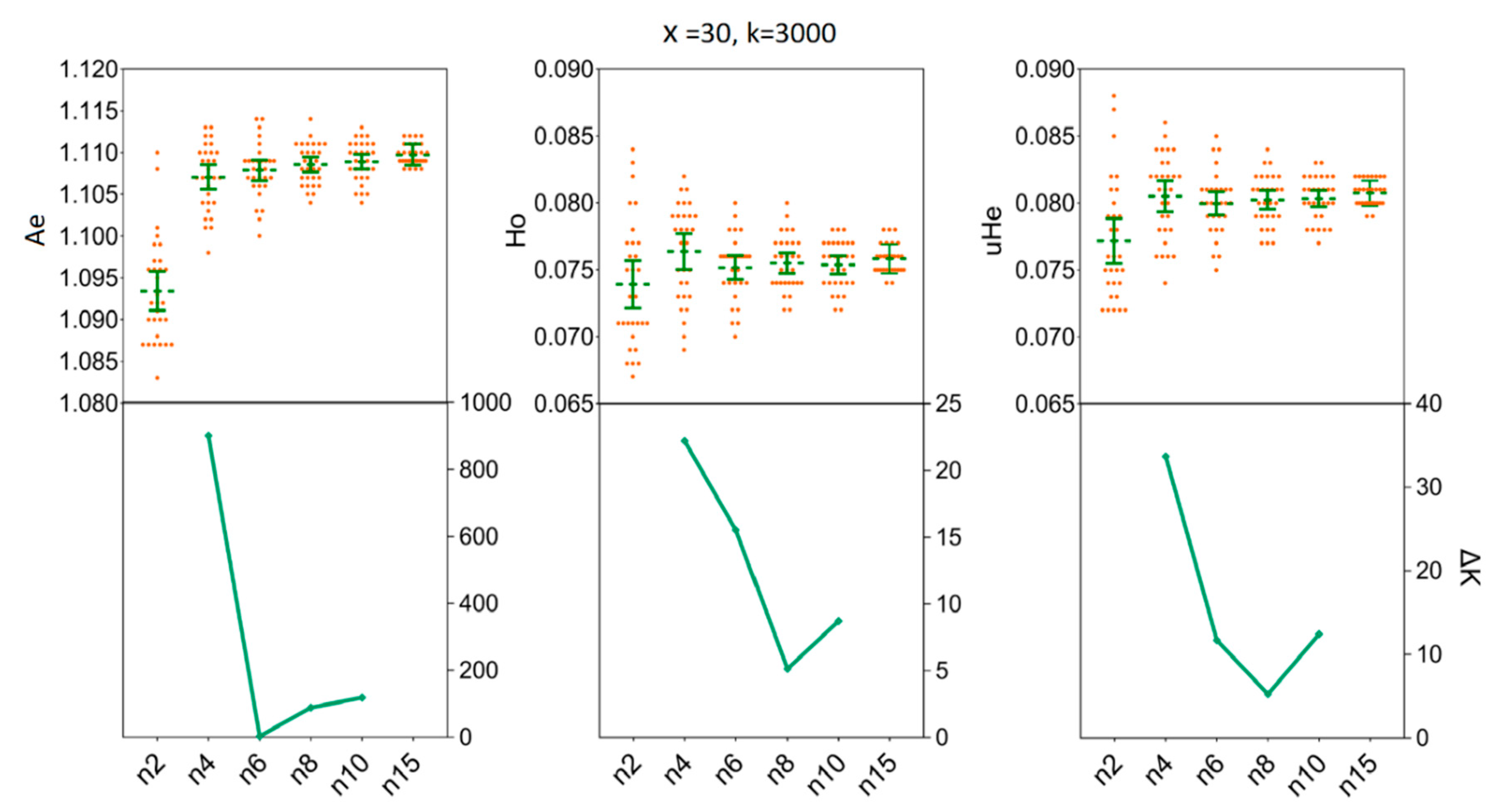
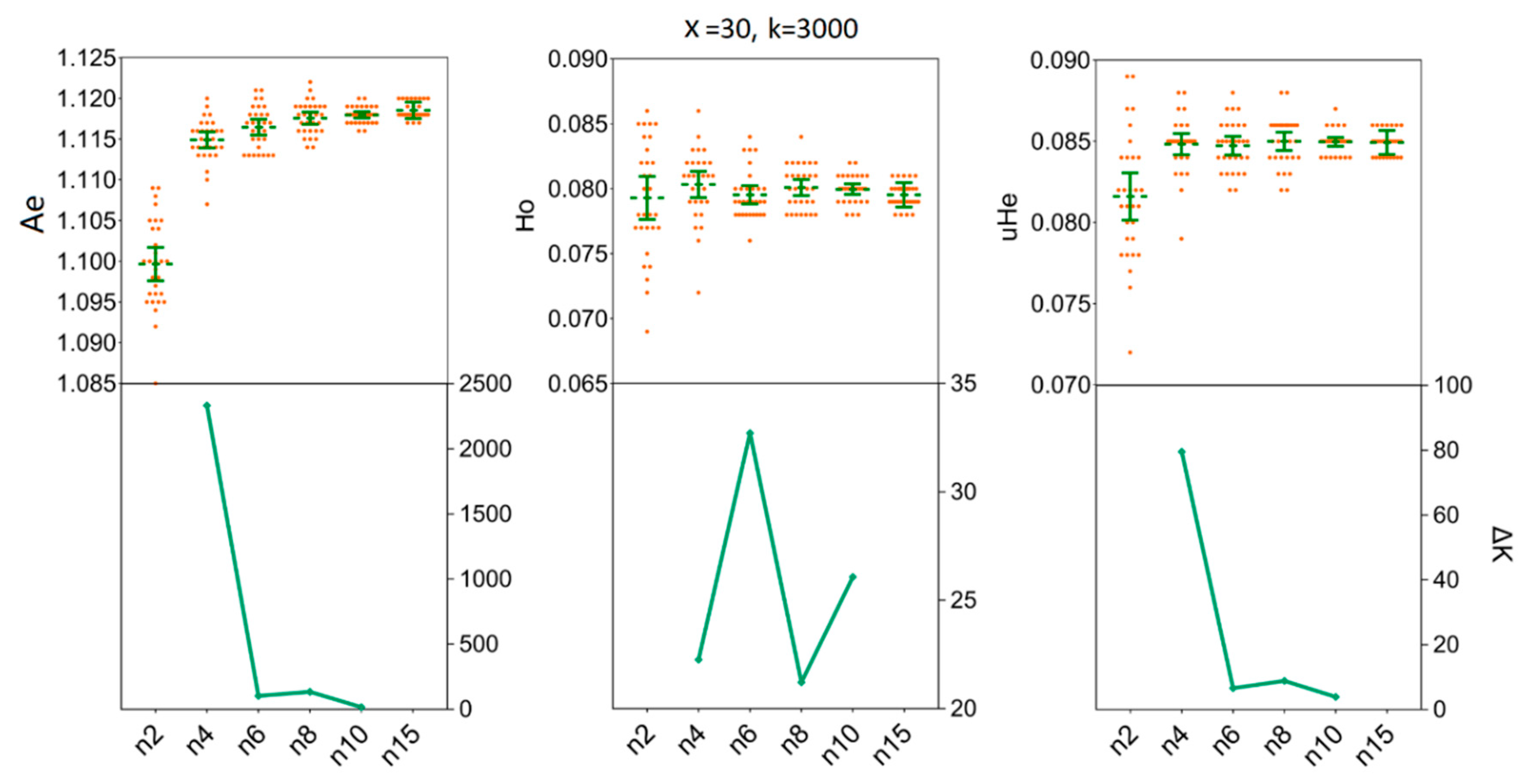
3.3. Depicting Sample Sizes for Intrapopulation Genetic Diversity
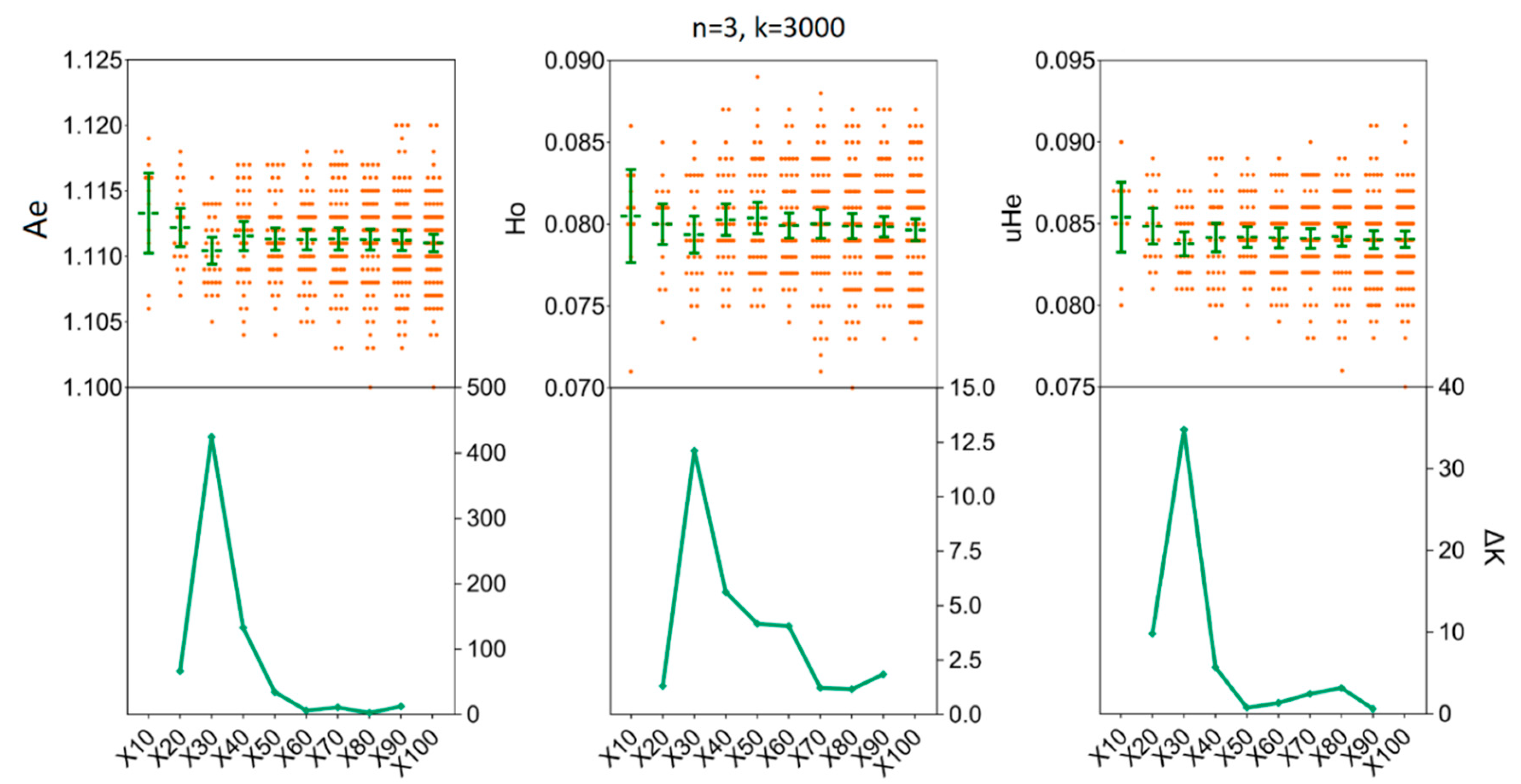
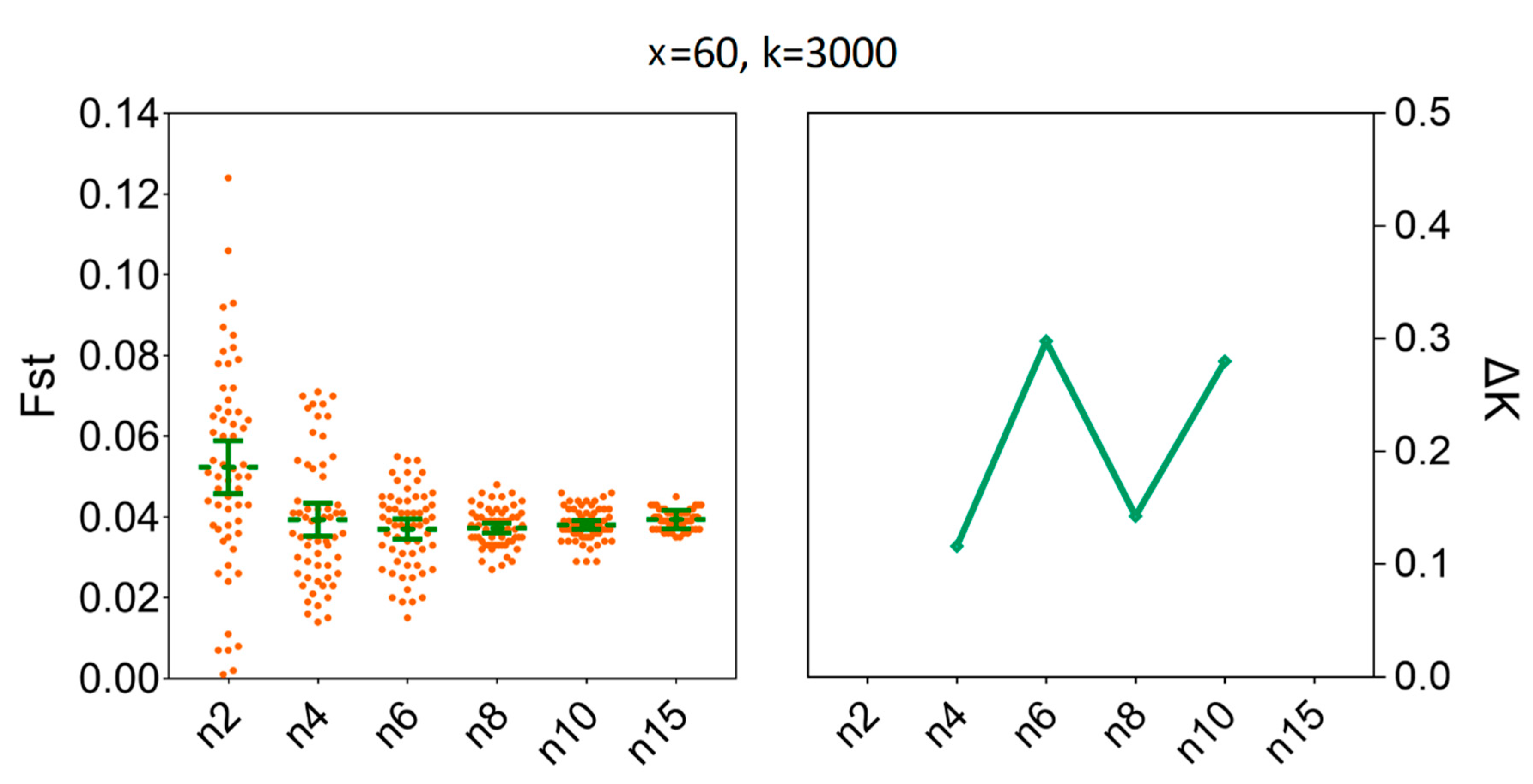
3.4. Determination of the Sample Sizes for Interpopulation Genetic Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Res. 2017, 17, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Willing, E.M.; Dreyer, C.; Oosterhout, C.V. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 2012, 7, e42649. [Google Scholar] [CrossRef] [PubMed]
- Swatdipong, A.; Primmer, C.; Vasemagi, A. Historical and recent genetic bottlenecks in European grayling, Thymallus thymallus. Conserv. Genet. 2010, 11, 279–292. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Jump, A.S. Species-genetic diversity correlations in habitat fragmentation can be biased by small sample sizes. Mol. Ecol. 2012, 21, 2847–2849. [Google Scholar] [CrossRef]
- Hobas, S.; Gaggiotti, O.; Consortium, C.; Bertorelle, G. Sample planning optimization tool for conservation and population genetics (SPOTG): A software for choosing the appropriate number of markers and samples. Methods Ecol. Evol. 2013, 4, 299–303. [Google Scholar]
- Paetkau, D.; Slade, R.; Burden, M.; Estoup, A. Genetic assignment methods for the direct, real-time estimation of migration rate: A simulation-based exploration of accuracy and power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef]
- Ward, S.M.; Jasieniuk, M. Review: Sampling weedy and invasive plant populations for genetic diversity analysis. Weed Sci. 2009, 57, 593–602. [Google Scholar] [CrossRef]
- Hale, M.L.; Burg, T.M.; Steeves, T.E. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS ONE 2012, 7, e45170. [Google Scholar] [CrossRef]
- Landguth, E.L.; Fedy, B.C.; Oyler-McCance, S.J.; Garey, A.L.; Emel, S.L.; Mumma, M.; Wanger, H.H.; Fortin, M.J.; Cushman, S.A. Effects of sample size, number of markers, and allelic richness on the detection of spatial genetic pattern. Mol. Ecol. Res. 2012, 12, 276–284. [Google Scholar] [CrossRef]
- Peery, M.Z.; Kirby, R.; Reid, B.N.; Stoelting, R.; Doucet-Bëer, E.; Robinson, S.; Vásquez-Carrillo, C.; Pauli, J.N.; Palsbøll, P.J. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012, 21, 3403–3418. [Google Scholar] [CrossRef]
- Lombaert, E.; Guillemaud, T.; Deleury, E. Biases of STRUCTURE software when exploring introduction routes of invasive species. Heredity 2018, 120, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Ralls, K.; Eldridge, M.D.B.; Dudash, M.R.; Fenster, C.B.; Lacy, R.C.; Sunnucks, P. Genetic Management of Fragmented Animal and Plant Populations; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Saura, M.; Fernández, A.; Rodríguez, M.C. Genome-wide estimates of coancestry and inbreeding in a closed herd of ancient iberian pigs. PLoS ONE 2013, 8, e78314. [Google Scholar] [CrossRef] [PubMed]
- Toro, M.A.; Villanueva, B.; Fernández, J. Genomics applied to management strategies in conservation programs. Livest. Sci. 2014, 166, 48–53. [Google Scholar] [CrossRef]
- Jeffries, D.L.; Copp, G.H.; Handley, L.L.; Olsén, H.K.; Sayer, C.D.; Hänfling, B. Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Biol. Invasions 2016, 25, 2997–3018. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef]
- Van Tassell, C.P.; Smith, T.P.; Matukumalli, L.K.; Taylor, J.F.; Schnabel, R.D.; Taylor Lawley, C.; Haudenschild, C.D.; Moore, S.S.; Warren, W.C.; Sonstegard, T.S. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 2008, 5, 247–252. [Google Scholar] [CrossRef]
- Deagle, B.E.; Faux, C.; Kawaguchi, S.; Meyer, B.; Jarman, S.N. Antarctic krill population genomics: Apparent panmixia, but genome complexity and large population size muddy the water. Mol. Ecol. 2015, 24, 4943–4959. [Google Scholar] [CrossRef]
- Martin, C.H.; Crawford, J.E.; Turner, B.J.; Turner, B.J.; Simons, L.H. Diabolical survival in Death Valley: Recent pupfish colonization, gene flow and genetic assimilation in the smallest species range on earth. Proc. Royal Soc. B 2016, 283, 20152334. [Google Scholar] [CrossRef]
- Ozerov, M.Y.; Gross, R.; Bruneaux, M.; Vähä, J.P.; Burimski, O.; Pukk, L.; Vasemägi, A. Genome-wide introgressive hybridization patterns in wild Atlantic salmon influenced by inadvertent gene flow from hatchery releases. Mol. Ecol. 2016, 25, 1275–1293. [Google Scholar] [CrossRef]
- Vera, M.; Díez-del-Molino, D.; García-Marín, J.L. Genomic survey provides insights into the evolutionary changes that occurred during European expansion of the invasive mosquitofish (Gambusia holbrooki). Mol. Ecol. 2016, 25, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.M.; Liang, N.; Wu, Z.K.; Zhao, Y.G.; Chu, D. Minimum sample sizes for invasion genomics: Empirical investigation in an invasive whitefly. Ecol. Evol. 2020, 10, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.L. The multicolored Asian lady beetle, Harmonia axyridis: A review of its biology, uses in biological control, and non-target impacts. J. Insect Sci. 2003, 3, 1–16. [Google Scholar] [CrossRef]
- Brown, P.M.J.; Thomas, C.E.; Lombaert, E.; Jeffries, D.L.; Estoup, A.; Handley, L.J.L. The global spread of Harmonia axyridis (Coleoptera: Coccinellidae): Distribution, dispersal and routes of invasion. Biocontrol 2011, 56, 623–641. [Google Scholar] [CrossRef]
- Van Lenteren, J.C. The state of commercial augmentative biological control: Plenty of natural enemies, but a frustrating lack of uptake. BioControl 2012, 57, 1–20. [Google Scholar] [CrossRef]
- Sloggett, J.J. Harmonia axyrids invasions: Deducing evolutionary causes and consequence. Entomol. Sci. 2012, 15, 261–273. [Google Scholar] [CrossRef]
- Roy, H.E.; Brown, P.M.J.; Adriaens, T.; Zhao, Z.H. The harlequin ladybird, Harmonia axyridis: Global perspectives on invasion history and ecology. Biol. Invasions 2016, 18, 997–1044. [Google Scholar] [CrossRef]
- Keller, S.R.; Taylor, D.R. History, chance and adaptation during biological invasion: Separating stochastic phenotypic evolution from response to selection. Ecol. Lett. 2008, 11, 852–866. [Google Scholar] [CrossRef]
- Simberloff, D.; Martin, J.L.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Franck, C.; Galil, B.; García-Berthou, E.; Pascal, M.; et al. Impacts of biological invasions: What’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef]
- Lombaert, E.; Guillemaud, T.; Lundgren, J.; Koch, R.; Facon, B.; Grez, A.; Loomans, A.; Malausa, T.; Nedved, O.; Rhule, E.; et al. Complementarity of statistical treatments to reconstruct worldwide routes of invasion: The case of the Asian ladybird Harmonia axyridis. Mol. Ecol. 2014, 23, 5979–5997. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meyer, E.; Mckay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Dou, J.Z.; Mao, J.X.; Su, H.L.; Jiao, W.Q.; Zhang, L.L.; Hu, X.L.; Huang, X.T.; Wang, S.; Bao, Z.M. RADtyping: An integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations. PLoS ONE 2013, 8, e79960. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Li, H.; Chen, Z.Z.; Coghlan, A.; Coin, L.J.M.; Guo, Y.R.; Hériché, J.; Hu, Y.F.; Kristiansen, K.; Li, R.Q.; et al. TreeFam: 2008 Update. Nucleic Acids Res. 2008, 36, 735–740. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Luikart, G.; Cornuet, J.M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Koskinen, M.T.; Hirvonen, H.; Landry, P.A.; Primmer, C.R. The benefits of increasing the number of microsatellites utilized in genetic populations studies: An empirical perspective. Hereditas 2004, 141, 61–67. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Do polymorphic loci require large sample sizes to estimate genetic distances? Heredity 2005, 94, 33–36. [Google Scholar] [CrossRef]
- González-Ramos, J.; Agell, G.; Uriz, M.J. Microsatellites from sponge genomes: The number necessary for detecting genetic structure in Hemimycale columella populations. Aquat. Biol. 2015, 24, 25–34. [Google Scholar] [CrossRef]
- Gaughran, S.J.; Quinzin, M.C.; Miller, M.J.; Garrick, R.C.; Edwards, D.L.; Russello, M.A.; Poulakakis, N.; Ciofi, C.; Beheregaray, L.B.; Caccone, A. Data from: Theory, practice, and conservation in the age of genomics: The Galápagos giant tortoise as a case study. Evol. Appl. 2017, 7, 1084–1093. [Google Scholar]
- Flesch, E.P.; Rotella, J.J.; Thomson, J.M.; Graves, T.A.; Garrott, R.A. Evaluating sample size to estimate genetic management metrics in the genomics era. Mol. Ecol. Res. 2018, 18, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, K.; Eeir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST). Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Lallias, D.; Boudry, P.; Batista, F.M.; Beaumont, A.; King, J.W.; Turner, J.R.; Lapègue, S. Invasion genetics of the Pacific oyster Crassostrea gigas, in the British Isles inferred from microsatellite and mitochondrial markers. Biol. Invasions 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Chu, D.; Qu, W.M.; Guo, L. Invasion genetics of alien insect pests in China: Research progress and future prospects. J. Integr. Agric. 2018, 18, 748–757. [Google Scholar] [CrossRef]
- Hoban, S.; Schlarbaum, S. Optimal sampling of seeds from plant populations for ex-situ conservation of genetic biodiversity, considering realistic population structure. Biol. Conserv. 2014, 177, 90–99. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Analyzed | Genetic Diversity | Genetic Differentiation | References | ||
|---|---|---|---|---|---|
| Ae | Ho | uHe | (Fst) | ||
| Bemisia tabaci MED Q1 clade | 3 | 3–4 | 3 | 4 | Qu et al. 2019 |
| Bemisia tabaci MED Q2 clade | 3–4 | 4 | 3–4 | 3 | Qu et al. 2019 |
| Amphirrhox longifolia | 2 | 2 | 6–8 | 2 | Nazareno et al. 2017 |
| Harmonia axyridis | 4 | 4–6 | 4 | 6 | The present study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Qu, W.; Obrycki, J.J.; Meng, L.; Zhou, X.; Chu, D.; Li, B. Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis. Insects 2020, 11, 290. https://doi.org/10.3390/insects11050290
Li H, Qu W, Obrycki JJ, Meng L, Zhou X, Chu D, Li B. Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis. Insects. 2020; 11(5):290. https://doi.org/10.3390/insects11050290
Chicago/Turabian StyleLi, Hongran, Wanmei Qu, John J. Obrycki, Ling Meng, Xuguo Zhou, Dong Chu, and Baoping Li. 2020. "Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis" Insects 11, no. 5: 290. https://doi.org/10.3390/insects11050290
APA StyleLi, H., Qu, W., Obrycki, J. J., Meng, L., Zhou, X., Chu, D., & Li, B. (2020). Optimizing Sample Size for Population Genomic Study in a Global Invasive Lady Beetle, Harmonia Axyridis. Insects, 11(5), 290. https://doi.org/10.3390/insects11050290