Development of a Tet-On Inducible Expression System for the Anhydrobiotic Cell Line, Pv11

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Vector Construction
2.3. Transfection and Luciferase Reporter Assay
2.4. Prediction of Core Promoter Elements
2.5. GFP Expression Using the Tet-On System
2.6. AMV RTα Expression Using the Tet-On System
2.7. Antibodies and Western Blot Analysis
2.8. Reverse Transcriptase Assay
2.9. Statistical Analysis
3. Results
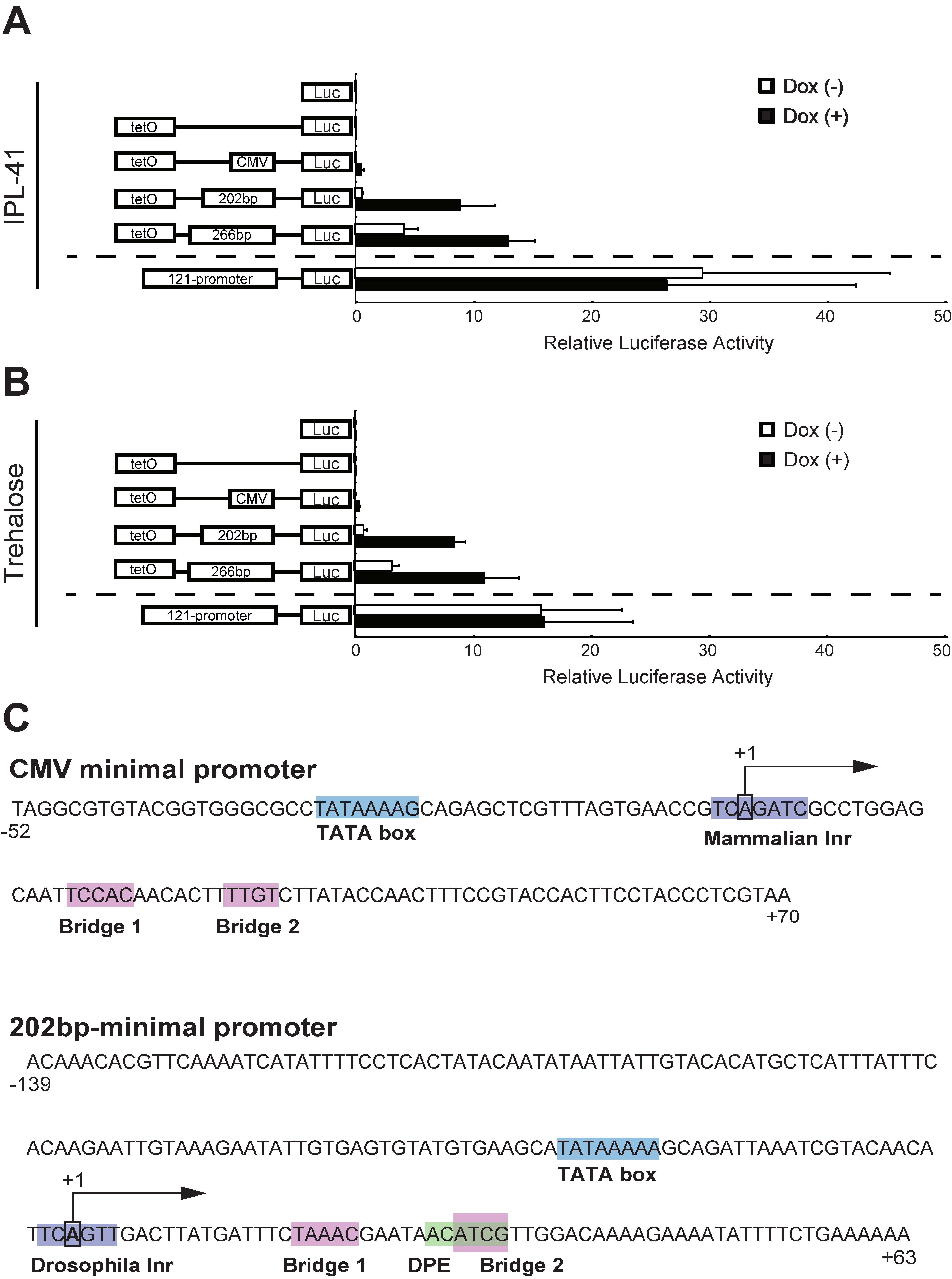
3.1. Identification of a Minimal Promoter for the Development of an Effective Inducible Expression System in Pv11 Cells Based on the Tet-On System
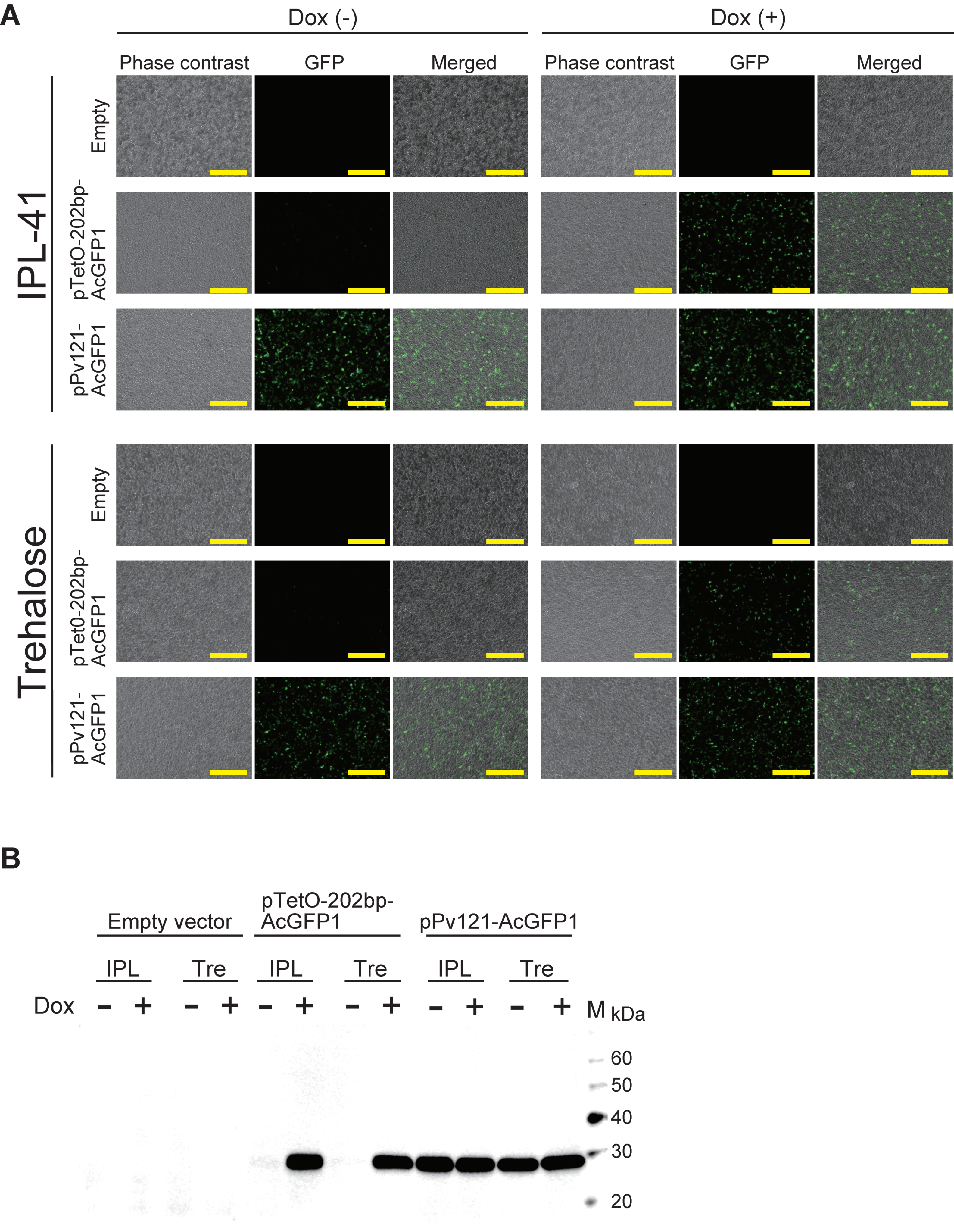
3.2. AcGFP1 Expression Using the Tet-On System in Pv11 Cells
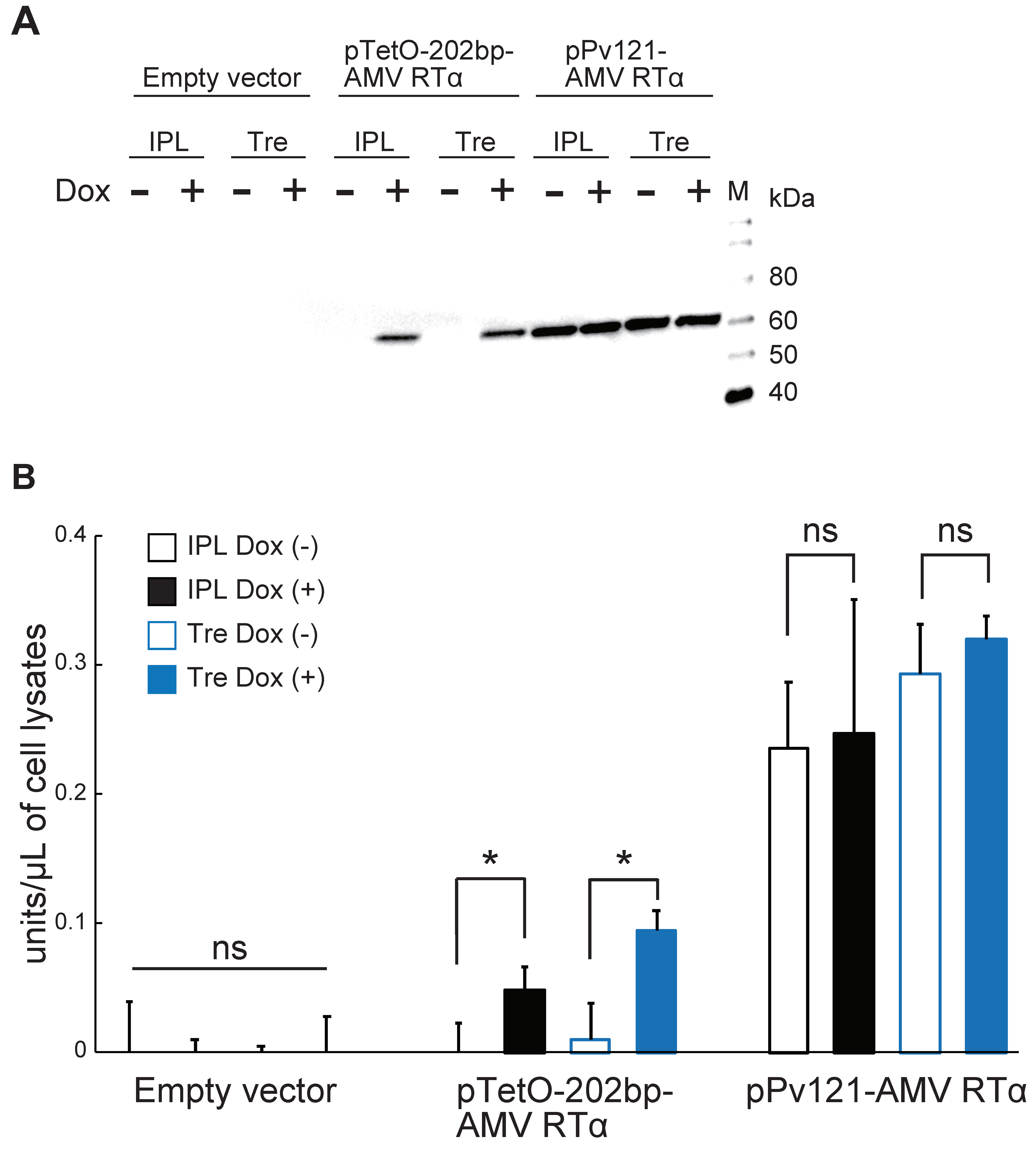
3.3. AMV RTα Expression and Measurement of RT Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brondyk, W.H. Selecting an Appropriate Method for Expressing a Recombinant Protein. Methods Enzymol. 2009, 463, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, M.M.; Pijlman, G.P.; Vlak, J.M. Thirty Years of Baculovirus–Insect Cell Protein Expression: From Dark Horse to Mainstream Technology. J. Gen. Virol. 2015, 96, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Felberbaum, R.S. The Baculovirus Expression Vector System: A Commercial Manufacturing Platform for Viral Vaccines and Gene Therapy Vectors. Biotechnol. J. 2015, 10, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Krey, T. Stable Drosophila Cell Lines: An Alternative Approach to Exogenous Protein Expression. Methods Mol. Biol. 2016, 1350, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Ardisson-Araújo, D.M.P.; Morgado, F.D.S.; Schwartz, E.F.; Corzo, G.; Ribeiro, B.M. A New theraphosid Spider Toxin Causes Early Insect Cell Death by Necrosis When Expressed In Vitro during Recombinant Baculovirus Infection. PLoS ONE 2013, 8, e84404. [Google Scholar] [CrossRef]
- Cox, M.M. Recombinant Protein Vaccines Produced in Insect Cells. Vaccine 2012, 30, 1759–1766. [Google Scholar] [CrossRef]
- Takahashi, M.; Mitsuhashi, J.; Ohtaki, T. Establishment of a Cell Line from Embryonic Tissues of the Fleshfly, Sarcophaga peregrina (Insecta, Diptera). Dev. Growth Differ. 1980, 22, 11–19. [Google Scholar] [CrossRef]
- Watanabe, K.; Imanishi, S.; Akiduki, G.; Cornette, R.; Okuda, T. Air-Dried Cells from the Anhydrobiotic Insect, Polypedilum vanderplanki, Can Survive Long Term Preservation at Room Temperature and Retain Proliferation Potential After Rehydration. Cryobiology 2016, 73, 93–98. [Google Scholar] [CrossRef]
- Cornette, R.; Kikawada, T. The Induction of Anhydrobiosis in the Sleeping Chironomid: Current Status of Our Knowledge. IUBMB Life 2011, 63, 419–429. [Google Scholar] [CrossRef]
- Crowe, J.H.; Crowe, L.M.; Oliver, A.E.; Tsvetkova, N.; Wolkers, W.; Tablin, F. The Trehalose Myth Revisited: Introduction to a Symposium on Stabilization of Cells in the Dry State. Cryobiology 2001, 43, 89–105. [Google Scholar] [CrossRef]
- Yamada, T.G.; Suetsugu, Y.; Deviatiiarov, R.; Gusev, O.; Cornette, R.; Nesmelov, A.; Hiroi, N.; Kikawada, T.; Funahashi, A. Transcriptome Analysis of the Anhydrobiotic Cell Line Pv11 Infers the Mechanism of Desiccation Tolerance and Recovery. Sci. Rep. 2018, 8, 17941. [Google Scholar] [CrossRef] [PubMed]
- Furuki, T.; Shimizu, T.; Chakrabortee, S.; Yamakawa, K.; Hatanaka, R.; Takahashi, T.; Kikawada, T.; Okuda, T.; Mihara, H.; Tunnacliffe, A.; et al. Effects of Group 3 LEA Protein Model Peptides on Desiccation-Induced Protein Aggregation. Biochim. Biophys. Acta 2012, 1824, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Furuki, T.; Sakurai, M. Group 3 LEA Protein Model Peptides Protect Liposomes during Desiccation. Biochim. Biophys. Acta 2014, 1838, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, S.; Watanabe, S.J.; Sato, R.; Gusev, O.; Nesmelov, A.; Sogame, Y.; Cornette, R.; Kikawada, T. Towards Water-Free Biobanks: Long-Term Dry-Preservation at Room Temperature of Desiccation-Sensitive Enzyme Luciferase in Air-Dried Insect Cells. Sci. Rep. 2017, 7, 6540. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Tokumoto, S.; Sogame, Y.; Deviatiiarov, R.; Okada, J.; Cornette, R.; Gusev, O.; Shagimardanova, E.; Sakurai, M.; Kikawada, T. Identification of a Novel Strong Promoter From the Anhydrobiotic Midge, Polypedilum vanderplanki, with Conserved Function in Various Insect Cell Lines. Sci. Rep. 2019, 9, 7004. [Google Scholar] [CrossRef]
- Dhanesuan, N.; Sharp, J.A.; Blick, T.; Price, J.T.; Thompson, E.W. Doxycycline-Inducible Expression of SPARC/Osteonectin/BM40 in MDA-MB-231 Human Breast Cancer Cells Results in Growth Inhibition. Breast Cancer Res. Treat. 2002, 75, 73–85. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Meyer, R.G.; Kaiser, H.; Brack, A.R.; Kandolf, R.; Küpper, J.-H. Comparative Analysis of Inducible Expression Systems in Transient Transfection Studies. Anal. Biochem. 2004, 334, 9–19. [Google Scholar] [CrossRef]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.-Z.; Lahn, B.T. Systematic Comparison of Constitutive Promoters and the Doxycycline-Inducible Promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems for Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef]
- Karasaki, N.; Mon, H.; Takahashi, M.; Lee, J.M.; Koga, K.; Kawaguchi, Y.; Kusakabe, T. Establishment of Tetracycline-Inducible Gene Expression Systems in the Silkworm, Bombyx Mori. Biotechnol. Lett. 2008, 31, 495–500. [Google Scholar] [CrossRef]
- Norrman, K.; Fischer, Y.; Bonnamy, B.; Sand, F.W.; Ravassard, P.; Semb, H. Quantitative Comparison of Constitutive Promoters in Human ES Cells. PLoS ONE 2010, 5, e12413. [Google Scholar] [CrossRef] [PubMed]
- Loew, R.; Heinz, N.; Hampf, M.; Bujard, H.; Gossen, M. Improved Tet- Responsive Promoters with Minimized Background Expression. BMC Biotechnol. 2010, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Danino, Y.M.; Even, D.; Ideses, D.; Juven-Gershon, T. The Core Promoter: At the Heart of Gene Expression. Biochim. Biophys. Acta. 2015, 1849, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Shir-Shapira, H.; Sloutskin, A.; Adato, O.; Ovadia-Shochat, A.; Ideses, D.; Zehavi, Y.; Kassavetis, G.; Kadonaga, J.T.; Unger, R.; Juven-Gershon, T. Identification of Evolutionarily Conserved Downstream Core Promoter Elements Required for the Transcriptional Regulation of Fushi Tarazu Target Genes. PLoS ONE 2019, 14, e0215695. [Google Scholar] [CrossRef]
- Theisen, J.W.M.; Lim, C.Y.; Kadonaga, J.T. Three Key Subregions Contribute to the Function of the Downstream RNA Polymerase II Core Promoter. Mol. Cell. Biol. 2010, 30, 3471–3479. [Google Scholar] [CrossRef]
- Even, D.Y.; Kedmi, A.; Basch-Barzilay, S.; Ideses, D.; Tikotzki, R.; Shir-Shapira, H.; Shefi, O.; Juven-Gershon, T. Engineered Promoters for Potent Transient Overexpression. PLoS ONE 2016, 11, e0148918. [Google Scholar] [CrossRef][Green Version]
- Nakahara, Y.; Imanishi, S.; Mitsumasu, K.; Kanamori, Y.; Iwata, K.-I.; Watanabe, M.; Kikawada, T.; Okuda, T. Cells from an Anhydrobiotic Chironomid Survive Almost Complete Desiccation. Cryobiology 2010, 60, 138–146. [Google Scholar] [CrossRef]
- Sogame, Y.; Okada, J.; Kikuta, S.; Miyata, Y.; Cornette, R.; Gusev, O.; Kikawada, T. Establishment of Gene Transfer and Gene Silencing Methods in a Desiccation-Tolerant Cell Line, Pv11. Extremophiles 2016, 21, 65–72. [Google Scholar] [CrossRef]
- Sloutskin, A.; Danino, Y.M.; Orenstein, Y.; Zehavi, Y.; Doniger, T.; Shamir, R.; Juven-Gershon, T. ElemeNT: A Computational Tool for Detecting Core Promoter Elements. Transcription 2015, 6, 41–50. [Google Scholar] [CrossRef]
- Pfeifer, T.; Hegedus, D.; Grigliatti, T.A.; Theilmann, D.A. Baculovirus Immediate-Early Promoter-Mediated Expression of the Zeocin™ Resistance Gene for Use as a Dominant Selectable Marker in Dipteran and Lepidopteran Insect Cell Lines. Gene 1997, 188, 183–190. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Liono, L.; Chen, S.-L.; Chen, C.Y.; Chao, Y.-C. Expression of Highly Controllable Genes in Insect Cells Using a Modified Tetracycline-Regulated Gene Expression System. J. Biotechnol. 2000, 80, 75–83. [Google Scholar] [CrossRef]
- Yamada, T.G.; Hiki, Y.; Hiroi, N.F.; Shagimardanova, E.; Gusev, O.; Cornette, R.; Kikawada, T.; Funahashi, A. Identification of a Master Transcription Factor and a Regulatory Mechanism for Desiccation Tolerance in the Anhydrobiotic Cell Line Pv11. PLoS ONE 2020, 15, e0230218. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Kimura, Y.; Lezhava, A.; Kanamori, H.; Usui, K.; Hanami, T.; Soma, T.; Morlighem, J.-É.; Saga, S.; Ishizu, Y.; et al. One-Step Detection of the 2009 Pandemic Influenza A(H1N1) Virus by the RT-SmartAmp Assay and Its Clinical Validation. PLoS ONE 2012, 7, e30236. [Google Scholar] [CrossRef] [PubMed]
- Kacian, D.; Watson, K.; Burny, A.; Spiegelman, S. Purification of the DNA Polymerase of Avian Myeloblastosis Virus. Biochim. Biophys. Acta 1971, 246, 365–383. [Google Scholar] [CrossRef]
- Konishi, A.; Nemoto, D.; Yasukawa, K.; Inouye, K. Comparison of the Thermal Stabilities of the Alphabeta Heterodimer and the Alpha Subunit of Avian Myeloblastosis Virus Reverse Transcriptase. Biosci. Biotechnol. Biochem. 2011, 75, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Hu, G.; Rickles, R.J.; Hannon, G.J.; Elledge, S.J. A Lentiviral MicroRNA-Based System for Single-Copy Polymerase II-Regulated RNA Interference in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13212–13217. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible In Vivo Genome Editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Nishijima, H.; Adachi, N.; Iiizumi, S.; Morohoshi, A.; Koyama, H.; Shibahara, K.-I. Generation of Tetracycline-Inducible Conditional Gene Knockout Cells in a Human Nalm-6 Cell Line. J. Biotechnol. 2009, 141, 1–7. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Zhang, J.; Wang, Y. Drug Inducible CRISPR/Cas Systems. Comput. Struct. Biotechnol. J. 2019, 17, 1171–1177. [Google Scholar] [CrossRef]
- Shin, K.-J.; Wall, E.A.; Zavzavadjian, J.R.; Santat, L.A.; Liu, J.; Hwang, J.-I.; Rebres, R.; Roach, T.; Seaman, W.; Simon, M.I.; et al. A Single Lentiviral Vector Platform for MicroRNA-Based Conditional RNA Interference and Coordinated Transgene Expression. Proc. Natl. Acad. Sci. USA 2006, 103, 13759–13764. [Google Scholar] [CrossRef]
- Muerdter, F.; Boryń, Ł.M.; Arnold, C.D. STARR-Seq—Principles and Applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Inoue, F.; Ahituv, N. Decoding Enhancers Using Massively Parallel Reporter Assays. Genomics 2015, 106, 159–164. [Google Scholar] [CrossRef] [PubMed]
- White, M.A. Understanding How Cis-Regulatory Function Is Encoded in DNA Sequence Using Massively Parallel Reporter Assays and Designed Sequences. Genomics 2015, 106, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ahituv, N. Gene Regulatory Elements, Major Drivers of Human Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F.; Mujacic, M. Recombinant Protein Folding and Misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging Concepts of Protein Folding In Vitro and In Vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Swaminathan, N. Biologically Active Reverse Transcriptases. Patent Application No. AU 26117/00A, 14 January 2000. [Google Scholar]
- Sobek, H.; Mueller, R.; Schmidt, M.; Frey, B.; Suppmann, B.; Schmuck, R.; Thalhofer, J.-P.; Pallua, P.; Pajatsch, M. Method for Produsing an Active Heterodimeric AMV-RT in Prokaryotic Cells. U.S. Patent 6,902,920B2, 7 June 2005. [Google Scholar]
- Miyata, Y.; Fuse, H.; Tokumoto, S.; Hiki, Y.; Deviatiiarov, R.; Yoshida, Y.; Yamada, T.G.; Cornette, R.; Gusev, O.; Shagimardanova, E.; et al. Cas9-mediated Genome Editing Reveals a Significant Contribution of Calcium Signaling Pathways to Anhydrobiosis in Pv11. bioRxiv 2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokumoto, S.; Miyata, Y.; Usui, K.; Deviatiiarov, R.; Ohkawa, T.; Kondratieva, S.; Shagimardanova, E.; Gusev, O.; Cornette, R.; Itoh, M.; et al. Development of a Tet-On Inducible Expression System for the Anhydrobiotic Cell Line, Pv11. Insects 2020, 11, 781. https://doi.org/10.3390/insects11110781
Tokumoto S, Miyata Y, Usui K, Deviatiiarov R, Ohkawa T, Kondratieva S, Shagimardanova E, Gusev O, Cornette R, Itoh M, et al. Development of a Tet-On Inducible Expression System for the Anhydrobiotic Cell Line, Pv11. Insects. 2020; 11(11):781. https://doi.org/10.3390/insects11110781
Chicago/Turabian StyleTokumoto, Shoko, Yugo Miyata, Kengo Usui, Ruslan Deviatiiarov, Takahiro Ohkawa, Sabina Kondratieva, Elena Shagimardanova, Oleg Gusev, Richard Cornette, Masayoshi Itoh, and et al. 2020. "Development of a Tet-On Inducible Expression System for the Anhydrobiotic Cell Line, Pv11" Insects 11, no. 11: 781. https://doi.org/10.3390/insects11110781
APA StyleTokumoto, S., Miyata, Y., Usui, K., Deviatiiarov, R., Ohkawa, T., Kondratieva, S., Shagimardanova, E., Gusev, O., Cornette, R., Itoh, M., Hayashizaki, Y., & Kikawada, T. (2020). Development of a Tet-On Inducible Expression System for the Anhydrobiotic Cell Line, Pv11. Insects, 11(11), 781. https://doi.org/10.3390/insects11110781