Characterization of the Complete Mitochondrial Genomes from Two Nitidulid Pests with Phylogenetic Implications

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and DNA Extraction
2.2. Sequence Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
3.1. Genome Organization
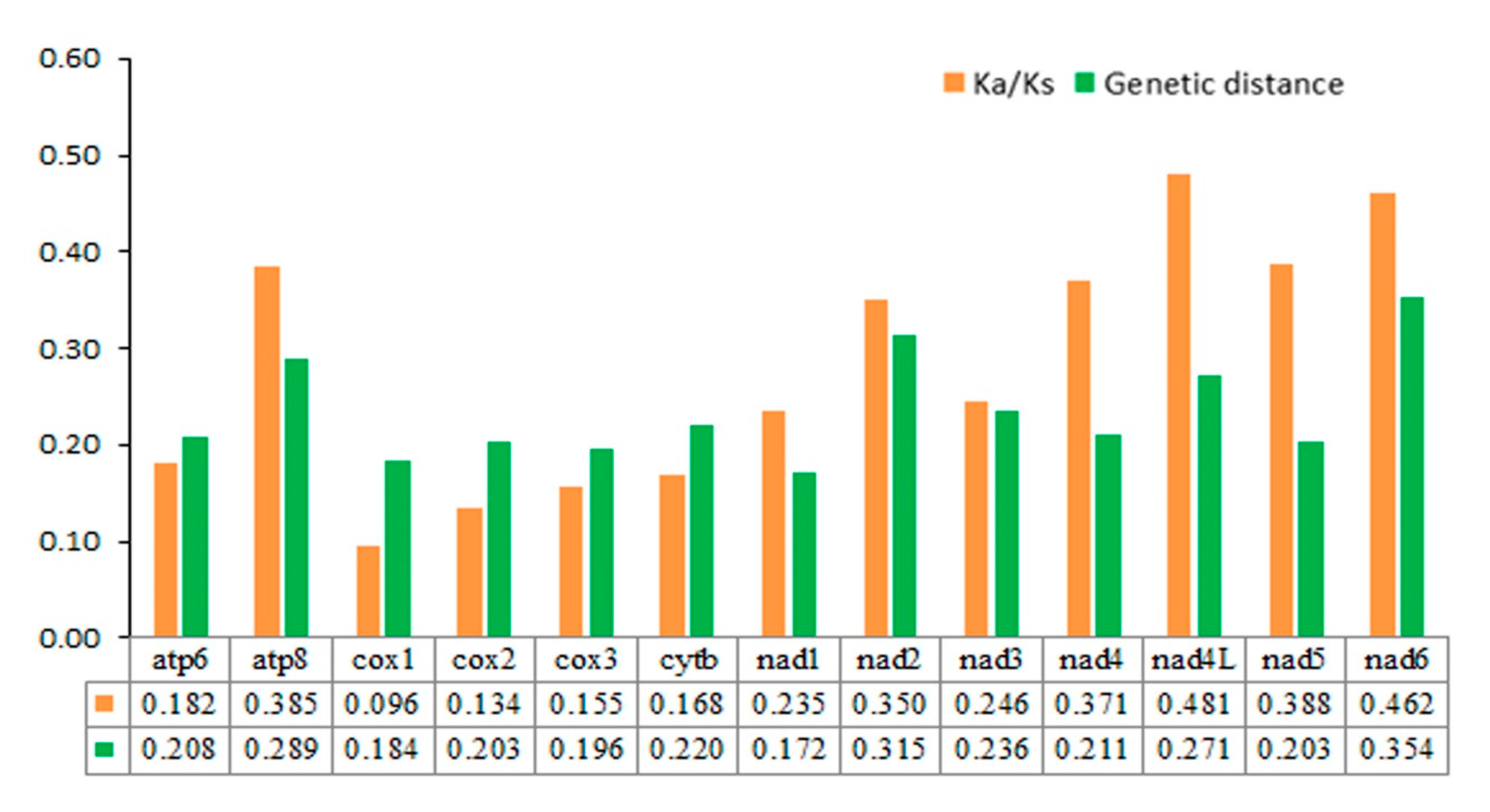
3.2. Protein-coding Genes (PCGs)
3.3. Transfer and Ribosomal RNAs
3.4. AT-rich Region
3.5. Nucleotide Analyses
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, M.H.; Lee, S.; Leschen, R.A.B.; Lee, S. Evolution of feeding habits of sap beetles (Coleoptera: Nitidulidae) and placement of Calonecrinae. Syst. Entomol. 2020, 45, 911–923. [Google Scholar] [CrossRef]
- Robertson, J.A.; Ślipinski, A.; Moulton, M.; Shockley, F.W.; Giorgi, A.; Lord, N.P.; McKenna, D.D.; Tomaszewska, W.; Forrester, J.; Miller, K.B.; et al. Phylogeny and classification of Cucujoidea and the recognition of a new superfamily Coccinelloidea (Coleoptera: Cucujiformia). Syst. Entomol. 2015, 40, 745–777. [Google Scholar] [CrossRef]
- Kirejtshuk, A.G. On recent knowledge on the sap-beetles (Coleoptera, Nitidulidae) of India. Zool. Inst. Russ. Acad. Sci. 1999, 1, 21–32. [Google Scholar]
- Audisio, P. Fauna d’Italia 32. Coleoptera, Nitidulidae–Kateridae; Edizioni Calderini: Roma, Italy, 1993; Volume 32, p. 971. [Google Scholar]
- Luo, F. Basic Research on One Kind of Pollination Beetles in Protected Field Agriculture-Haptoncus luteolus. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2004. [Google Scholar]
- Parsons, C.T. A revision of Nearctic Nitidulidae (Coleoptera). Bull. Mus. Comp. Zool. 1943, 92, 121–278. [Google Scholar]
- Lounsberry, Z.; Spiewok, S.; Pernal, S.F.; Sonstegard, T.S.; Hood, W.M.; Pettis, J.; Neumann, P.; Evans, J.D. Worldwide diaspora of Aethina tumida (Coleoptera: Nitidulidae), a nest parasite of honey bees. Ann. Entomol. Soc. Am. 2010, 103, 671–677. [Google Scholar] [CrossRef]
- Krishnan, K.T.; Neumann, P.; Ahmad, A.H.; Pimid, M. A scientific note on the association of Haptoncus luteolus (Coleoptera: Nitidulidae) with colonies of multiple stingless bee species. Apidologie 2014, 46, 262–264. [Google Scholar] [CrossRef]
- Zhao, H.X.; Chen, D.F.; Hou, C.S.; Wang, H.T.; Huang, W.Z.; Ji, C.H.; Ren, Q.; Xia, X.S.; Zhang, X.F. Biological characteristics of beehive beetle: Invasion hazard and its prevention and control countermeasures. J. Bee 2019, 1, 8–11. [Google Scholar]
- Dobson, R.M. The species of Carpophilus Stephens (Coleoptera: Nitidulidae) associated with stored products. Bull. Entomol. Res. 1954, 45, 389–402. [Google Scholar] [CrossRef]
- Nadel, H.; Pena, J.E. Identity, behavior, and efficacy of nitidulid beetles (Coleoptera: Nitidulidae) pollinating commercial Annona species in Florida. Environ. Entomol. 1994, 23, 878–886. [Google Scholar] [CrossRef]
- Glinski, Z.; Kostro, K.; Klimek, E. Aethina tumida-parasite and scavenger of the honeybee. Med. Weter. 2001, 57, 315–317. [Google Scholar]
- Deng, G.R.; Zeng, D.Q.; Li, W.Q.; Huang, D.X.; Zhou, Z.H. The harm of Tricanus japonicus on Dictyophora indusiata and its control. Plant Prot. 2006, 32, 117–118. [Google Scholar]
- Liao, T.L.; Yang, X.J.; An, Y.L.; Li, B.S.; Zhou, R. Several important Nitidulidae that transmit blight in the American oak. Plant Prot. 2005, 19, 37–38. [Google Scholar]
- Fan, H.S. A new record storage nitidulid and key for related species from China. J. Chin. Cereals Oils Assoc. 1993, 8, 1–3. [Google Scholar]
- Bai, X.G.; Cao, Y.; Zhou, Y.X. Identification of seven species of larvae of Nitidulidae from China. J. Zhengzhou Grain Coll. 1992, 2, 50–56. [Google Scholar]
- Zhou, Y.X.; Hao, L.J. A description of four species of storage Nitidulidae. J. Zhengzhou Grain Coll. 1997, 18, 99–100. [Google Scholar]
- Nie, W.G. Investigation and Classification of Stored Pests in Heilongjiang Province. Master’s Thesis, Northeast Agricultural University, Heilongjiang, China, 2004. [Google Scholar]
- He, C.G.; Wang, G.L.; Fan, Y.H.; Zou, Y.X.; Deng, Y.X.; Deng, H.Y. Study on a new pest of oilseed-Xenostrongylus variegatus. Acta Agric. Boreali Occident. Sin. 1998, 7, 18–23. [Google Scholar]
- Yang, Q.B.; Liu, W.C.; Huang, C. Statistics and analysis on oilseed rape losses caused by main diseases and insect pests in recent ten years. Plant Prot. 2018, 44, 24–30. [Google Scholar]
- Cline, A.R.; Smith, T.R.; Miller, K.; Moulton, M.; Whiting, M.; Audisio, P. Molecular phylogeny of Nitidulidae: Assessment of subfamilial and tribal classification and formalization of the family Cybocephalidae (Coleoptera: Cucujoidea). Syst. Entomol. 2014, 39, 758–772. [Google Scholar] [CrossRef]
- Lawrence, J.F.; Slipi’nski, A.; Seago, A.; Thayer, M.; Newton, A.; Marvaldi, A. Phylogeny of the Coleoptera based on adult and larval morphology. Ann. Zool. 2011, 61, 1–217. [Google Scholar] [CrossRef]
- Tang, P.; Li, M.; Feng, R.; Wang, J.; Liu, M.; Wang, Y.; Yuan, M. Phylogenetic relationships among superfamilies of Cucujiformia (Coleoptera: Polyphaga) inferred from mitogenomic data. Sci. Sinca 2019, 39, 163–171. [Google Scholar] [CrossRef]
- Bocak, L.; Barton, C.; Crampton-Platt, A.; Chesters, D.; Ahrens, D.; Vogler, A.P. Building the Coleoptera tree-of-life for >8000 species: Composition of public DNA data and fit with Linnaean classification. Syst. Entomol. 2014, 39, 97–110. [Google Scholar] [CrossRef]
- Leschen, R.A.B.; Lawrence, J.F.; Ślipiński, S.A. Classification of basal Cucujoidea (Coleoptera:Polyphaga): Cladistic analysis, keys and review of new families. Invertebr. Syst. 2005, 19, 17. [Google Scholar] [CrossRef]
- Jelínek, J.; Carlton, C.; Cline, A.R.; Leschen, R.A.B. Nitidulidae Latreille, 1802. In Handbuch der Zoologie/Handbook of Zoology; Leschen, R.A.B., Beutel, R.G., Lawrence, J.F., Eds.; Band/Vol. IV: Arthropoda: Insecta Teilband/Part 38. Coleoptera, Beetles. Morphology and Systematics (Polyphaga partim); W. De Gruyter: Berlin, Germany, 2010; Volume 2, pp. 390–407. [Google Scholar]
- Cline, A.R.; Slipinski, S.A. Discolomatidae Horn, 1878. In Handbuch der Zoologie/Handbook of Zoology; Leschen, R.A.B., Beutel, R.G., Lawrence, J.F., Eds.; Band. Arthropoda: Insecta Teilband; W. DeGruyter: Berlin, Germany, 2010; Volume IV, pp. 435–442. [Google Scholar]
- Hunt, T.; Bergsten, J.; Levkanicova, Z.; Papadopoulou, A.; John, O.S.; Wild, R.; Hammond, P.M.; Ahrens, D.; Balke, M.; Caterino, M.S.; et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Linard, B.; Crampton-Platt, A.; Moriniere, J.; Timmermans, M.J.T.N.; Andújar, C.; Arribas, P.; Miller, K.E.; Lipecki, J.; Favreau, E.; Hunter, A.; et al. The contribution of mitochondrial metagenomics to large-scale data mining and phylogenetic analysis of Coleoptera. Mol. Phylogenet. Evol. 2018, 128, 1–11. [Google Scholar] [CrossRef]
- Crampton-Platt1, A.; Timmermans, M.J.T.N.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Khen, C.V.; Vogler, A.P. Soup to tree: The phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef] [PubMed]
- Duquesne, V.; Delcont, A.; Huleux, A.; Beven, V.; Touzain, F.; Ribière-Chabert, M. Complete mitochondrial genome sequence of Aethina tumida (Coleoptera: Nitidulidae), a beekeeping pest. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lan, Y.; Xia, L.; Cui, M.; Sun, W.; Dong, Z.; Cao, Y. The first complete mitochondrial genomes of two sibling species from Nitidulid beetles pests. Insects 2019, 11, 24. [Google Scholar] [CrossRef]
- Song, H.; Sheffield, N.C.; Cameron, S.L.; Miller, K.B.; Whiting, M.F. When phylogenetic assumptions are violated: Base compositional heterogeneity and among-site rate variation in beetle mitochondrial phylogenomics. Syst. Entomol. 2010, 35, 429–448. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genet. Soc. Am. 1993, 133, 97–117. [Google Scholar]
- Shao, R.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003, 20, 362–370. [Google Scholar] [CrossRef]
- Zhang, S.K.; Shu, J.P.; Wang, Y.D.; Liu, Y.N.; Peng, H.; Zhang, W.; Wang, H.J. The complete mitochondrial genomes of two sibling species of camellia weevils (Coleoptera: Curculionidae) and patterns of Curculionini speciation. Sci. Rep. 2019, 9, 3412. [Google Scholar] [CrossRef]
- Yamauchi, M.M.; Miya, M.U.; Nishida, M. Use of a PCR-based approach for sequencing whole mitochondrial genomes of insects: Two examples (cockroach and dragonfly) based on the method developed for decapod crustaceans. Insect Mol. Biol. 2004, 13, 435–442. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Yang, D. The first mitochondrial genome for caddisfly (Insecta: Trichoptera) with phylogenetic implications. Int. J. Biol. Sci. 2013, 10, 53–63. [Google Scholar] [CrossRef]
- Wang, J.J.; Li, D.F.; Li, H.; Yang, M.F.; Dai, R.H. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci. Rep. 2019, 9, 15746. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Mardulyn, P.; Termonia, A.; Milinkovitch, M.C. Structure and evolution of the mitochondrial control region of leaf beetles (Coleoptera: Chrysomelidae): A hierarchical analysis of nucleotide sequence variation. J. Mol. Evol. 2003, 56, 38–45. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Accession Number | Reference |
|---|---|---|---|
| Sphindidae | Aspidiphorus orbiculatus | KT780625 | Unpublished |
| Erotylidae | Languriidae sp. | MG193464 | [36] |
| Erotylinae sp1 | MH836601 | [37] | |
| Erotylinae sp2 | MH789736 | [37] | |
| Monotomidae | Monotoma quadricollis | KX035132 | Unpublished |
| Rhizophagus aeneus | KX087340 | Unpublished | |
| Kateretidae | Brachypterolus vestitus | KX087245 | Unpublished |
| Nitidulidae | Nitidulidae sp | MH789742 | [37] |
| Aethina tumida | NC_036104 | [38] | |
| Xenostrongylusvariegatus | MW044620 | This study | |
| Epuraea guttata | KX087289 | Unpublished | |
| Carpophilus dimidiatus | NC_046036 | [39] | |
| Carpophilus pilosellus | MN604383 | [39] | |
| Epuraea sp. | MW044619 | This study | |
| Silvanidae | Uleiota sp. | KX035149 | Unpublished |
| Cucujidae | Cucujus clavipes | GU176341 | [40] |
| Cucujus haematodes | KX087268 | Unpublished |
| Regions | Size (bp) | T(U) | C | A | G | AT(%) | GC(%) | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|---|---|
| X. variegatus | |||||||||
| Full genome | 17,657 | 37.8 | 13 | 39.4 | 9.8 | 77.2 | 22.8 | 0.021 | −0.141 |
| PCGs | 11,046 | 43 | 11.5 | 34 | 11.5 | 77 | 23 | −0.116 | 0 |
| 1st codon position | 3682 | 37.2 | 10.7 | 35.1 | 17.1 | 72.3 | 27.8 | −0.029 | 0.229 |
| 2nd codon position | 3682 | 47.3 | 17.7 | 21.6 | 13.4 | 68.9 | 31.1 | −0.374 | −0.136 |
| 3rd codon position | 3682 | 44.4 | 6.2 | 45.4 | 4 | 89.8 | 10.2 | 0.012 | −0.207 |
| tRNAs | 1454 | 38.6 | 9 | 39.6 | 12.8 | 78.2 | 21.8 | 0.013 | 0.174 |
| rRNAs | 2079 | 42.8 | 6.6 | 38.5 | 12.1 | 81.3 | 18.7 | −0.053 | 0.296 |
| AT-rich region | 2910 | 40.2 | 13.5 | 34.4 | 11.9 | 74.6 | 25.4 | −0.078 | −0.064 |
| Epuraea sp. | |||||||||
| Full genome | 16,641 | 38.8 | 14.4 | 37.6 | 9.3 | 76.4 | 23.7 | −0.015 | −0.216 |
| PCGs | 11,097 | 42.9 | 12.9 | 32 | 12.2 | 74.9 | 25.1 | −0.146 | −0.026 |
| 1st codon position | 3699 | 36.7 | 11.9 | 34.4 | 17 | 71.1 | 28.9 | −0.032 | 0.179 |
| 2nd codon position | 3699 | 46.7 | 18.3 | 21.2 | 13.7 | 67.9 | 32 | −0.376 | −0.143 |
| 3rd codon position | 3699 | 45.2 | 8.5 | 40.3 | 6 | 85.5 | 14.5 | −0.057 | −0.175 |
| tRNAs | 1445 | 36.3 | 10.9 | 39.4 | 13.4 | 75.7 | 24.3 | 0.041 | 0.103 |
| rRNAs | 2081 | 41.9 | 6.9 | 36.9 | 14.4 | 78.8 | 21.3 | −0.063 | 0.353 |
| AT-rich region | 1984 | 53.8 | 12.1 | 28.8 | 5.2 | 82.6 | 17.3 | −0.302 | −0.397 |
| Species | Whole Genome | AT Skew | GC Skew | PCGs | tRNAs | rRNAs | A + T-Rich Region | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | AT (%) | Size (bp) | AT (%) | Size (bp) | AT (%) | Size (bp) | AT (%) | Size (bp) | AT (%) | |||
| E1 | 16,021 | 76.5 | 0.043 | −0.19 | 11,073 | 75.7 | 1451 | 75.7 | 2081 | 76.4 | - | - |
| E2 | 16,641 | 76.4 | −0.015 | −0.216 | 11,097 | 74.9 | 1445 | 75.8 | 2081 | 78.8 | 1984 | 82.6 |
| C1 | 15,717 | 75.2 | 0.038 | −0.202 | 11,094 | 74.5 | 1441 | 74.9 | 2061 | 75 | 1057 | 83.6 |
| C2 | 15,686 | 77.2 | 0.027 | −0.177 | 11,103 | 76.5 | 1442 | 76.5 | 2079 | 77.5 | 944 | 86.7 |
| N | 17,432 | 78.4 | 0.036 | −0.183 | 11,091 | 76.3 | 1443 | 78.2 | 2073 | 80.3 | - | - |
| A | 16,576 | 76.9 | 0.034 | −0.223 | 11,109 | 75.4 | 1460 | 77.2 | 2064 | 79.5 | - | - |
| X | 17,657 | 77.2 | 0.021 | −0.141 | 11,046 | 77 | 1454 | 78.2 | 2079 | 81.3 | 2910 | 74.6 |
| Position | Size (bp) | Intergenic Nucleotides | Codon | Strand | |||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| X. variegatus/E. sp. | |||||||
| trnI | 1/1 | 64/63 | 64/63 | +/+ | |||
| trnQ | 62/61 | 130/129 | 69/69 | −3/−3 | −/− | ||
| trnM | 131/129 | 199/197 | 69/69 | /−1 | +/+ | ||
| nad2 | 200/198 | 1174/1205 | 975/1008 | ATT/ATT | TAA/TAA | +/+ | |
| trnW | 1202/1214 | 1268/1280 | 67/67 | 27/8 | +/+ | ||
| trnC | 1383/1284 | 1446/1345 | 64/62 | 114/3 | −/− | ||
| trnY | 1448/1346 | 1510/1410 | 63/65 | 1/ | −/− | ||
| cox1 | 1503/1403 | 3042/2942 | 1540/1540 | −8/−8 | ATT/ATC | T/T | +/+ |
| trnL2 | 3043/2943 | 3107/3007 | 65/65 | +/+ | |||
| cox2 | 3108/3008 | 3780/3695 | 673/688 | ATT/ATT | T/T | +/+ | |
| trnK | 3781/3696 | 3851/3765 | 71/70 | +/+ | |||
| trnD | 3855/3766 | 3924/3831 | 70/66 | 3/ | +/+ | ||
| atp8 | 3925/3832 | 4069/3987 | 145/156 | ATC/ATC | T/TAG | +/+ | |
| atp6 | 4076/3981 | 4747/4655 | 672/675 | 6/−7 | ATA/ATG | TAA/TAA | +/+ |
| cox3 | 4747/4655 | 5533/5438 | 787/784 | −1/−1 | ATG/ATG | T/TAG | +/+ |
| trnG | 5534/5439 | 5597/5501 | 64/63 | +/+ | |||
| nad3 | 5604/5502 | 5951/5855 | 348/354 | 6/ | ATT/ATT | TAG/T | +/+ |
| trnA | 5950/5854 | 6015/5917 | 66/64 | −2/−2 | +/+ | ||
| trnR | 6015/5918 | 6077/5979 | 63/62 | −1/ | +/+ | ||
| trnN | 6077/5980 | 6142/6046 | 66/67 | −1/ | +/+ | ||
| trnS1 | 6143/6047 | 6209/6113 | 67/67 | +/+ | |||
| trnE | 6210/6114 | 6273/6176 | 64/63 | +/+ | |||
| trnF | 6272/6175 | 6336/6239 | 65/65 | −2/−2 | −/− | ||
| nad5 | 6337/6249 | 8053/7953 | 1717/1705 | /9 | ATA/ATT | T/TAG | −/− |
| trnH | 8051/7954 | 8114/8018 | 64/65 | −3/ | −/− | ||
| nad4 | 8112/8016 | 9444/9342 | 1333/1327 | −3/−3 | ATT/ATA | T/T | −/− |
| nad4L | 9435/9339 | 9722/9623 | 288/285 | −10/−4 | ATG/ATG | TAA/TAA | −/− |
| trnT | 9725/9626 | 9789/9689 | 65/64 | 2/2 | +/+ | ||
| trnP | 9790/9690 | 9854/9755 | 65/66 | −/− | |||
| nad6 | 9859/9760 | 10,359/10,263 | 501/504 | 4/4 | ATA/ATA | TAA/TAA | +/+ |
| cytb | 10,359/10,263 | 11,498/11,405 | 1140/1143 | −1/−1 | ATG/ATG | TAG/TAG | +/+ |
| trnS2 | 11,497/11,404 | 11,564/11,471 | 68/68 | −2/−2 | +/+ | ||
| nad1 | 11,582/11,489 | 12,514/12,421 | 933/933 | 17/17 | ATT/ATT | TAG/TAG | −/− |
| trnL1 | 12,534/12,441 | 12,600/12,505 | 67/65 | 19/19 | −/− | ||
| rrnL | 12,601/12,506 | 13,891/13,805 | 1291/1300 | −/− | |||
| trnV | 13,892/13,806 | 13,959/13,875 | 68/70 | −/− | |||
| rrnS | 13,960/13,877 | 14,747/14,657 | 788/781 | /1 | −/− | ||
| AT-rich region | 14,748/14,658 | 17,657/16,641 | 2910/1984 | +/+ | |||
| Gene | Start Codon/Stop Codon | ||||||
|---|---|---|---|---|---|---|---|
| E1 | E2 | C1 | C2 | N | A | X | |
| nad2 | ATT/TAA | ATT/TAA | ATT/TAA | ATT/TAA | ATT/T | ATT/TAA | ATT/TAA |
| cox1 | ATT/T | ATC/T | ATT/T | ATT/T | ATT/T | ATA/T | ATT/T |
| cox2 | ATA/T | ATT/T | ATC/T | ATT/T | ATT/TAG | ATT/T | ATT/T |
| atp8 | ATT/TAG | ATC/TAG | ATC/TAG | ATC/TAG | ATG/TAA | ATT/TAG | ATC/T |
| atp6 | ATG/TAA | ATG/TAA | ATG/TAA | ATA/TAA | ATG/TAA | ATA/TAA | ATA/TAA |
| cox3 | ATG/T | ATG/T | ATG/T | ATG/T | ATT/TAA | ATG/T | ATG/T |
| nad3 | ATA/TAG | ATT/TAG | ATT/TAG | ATT/TAG | ATT/TAA | ATA/TAG | ATT/TAG |
| nad5 | ATA/T | ATT/T | ATT/T | ATT/T | TAG/TAA | ATA/T | ATA/T |
| nad4 | ATG/TAA | ATA/T | ATG/T | ATG/T | ATG/TAA | ATG/T | ATT/T |
| nad4L | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATT/TAA | ATG/TAA | ATG/TAA |
| nad6 | ATC/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATG/TAG | ATA/TAA | ATA/TAA |
| Cytb | ATA/TAG | ATG/TAG | ATG/TAG | ATG/TAG | TTG/TAG | ATG/TAA | ATG/TAG |
| nad1 | AAC/ATC | ATT/TAG | ATA/TAG | ATG/TAG | ATT/TAA | TTG/TAG | ATT/TAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Song, Q.; Huang, M. Characterization of the Complete Mitochondrial Genomes from Two Nitidulid Pests with Phylogenetic Implications. Insects 2020, 11, 779. https://doi.org/10.3390/insects11110779
Chen X, Song Q, Huang M. Characterization of the Complete Mitochondrial Genomes from Two Nitidulid Pests with Phylogenetic Implications. Insects. 2020; 11(11):779. https://doi.org/10.3390/insects11110779
Chicago/Turabian StyleChen, Xiaoxiao, Qing Song, and Min Huang. 2020. "Characterization of the Complete Mitochondrial Genomes from Two Nitidulid Pests with Phylogenetic Implications" Insects 11, no. 11: 779. https://doi.org/10.3390/insects11110779
APA StyleChen, X., Song, Q., & Huang, M. (2020). Characterization of the Complete Mitochondrial Genomes from Two Nitidulid Pests with Phylogenetic Implications. Insects, 11(11), 779. https://doi.org/10.3390/insects11110779