Competitive Displacement between Bemisia tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED


Abstract
1. Introduction
2. Materials and Methods
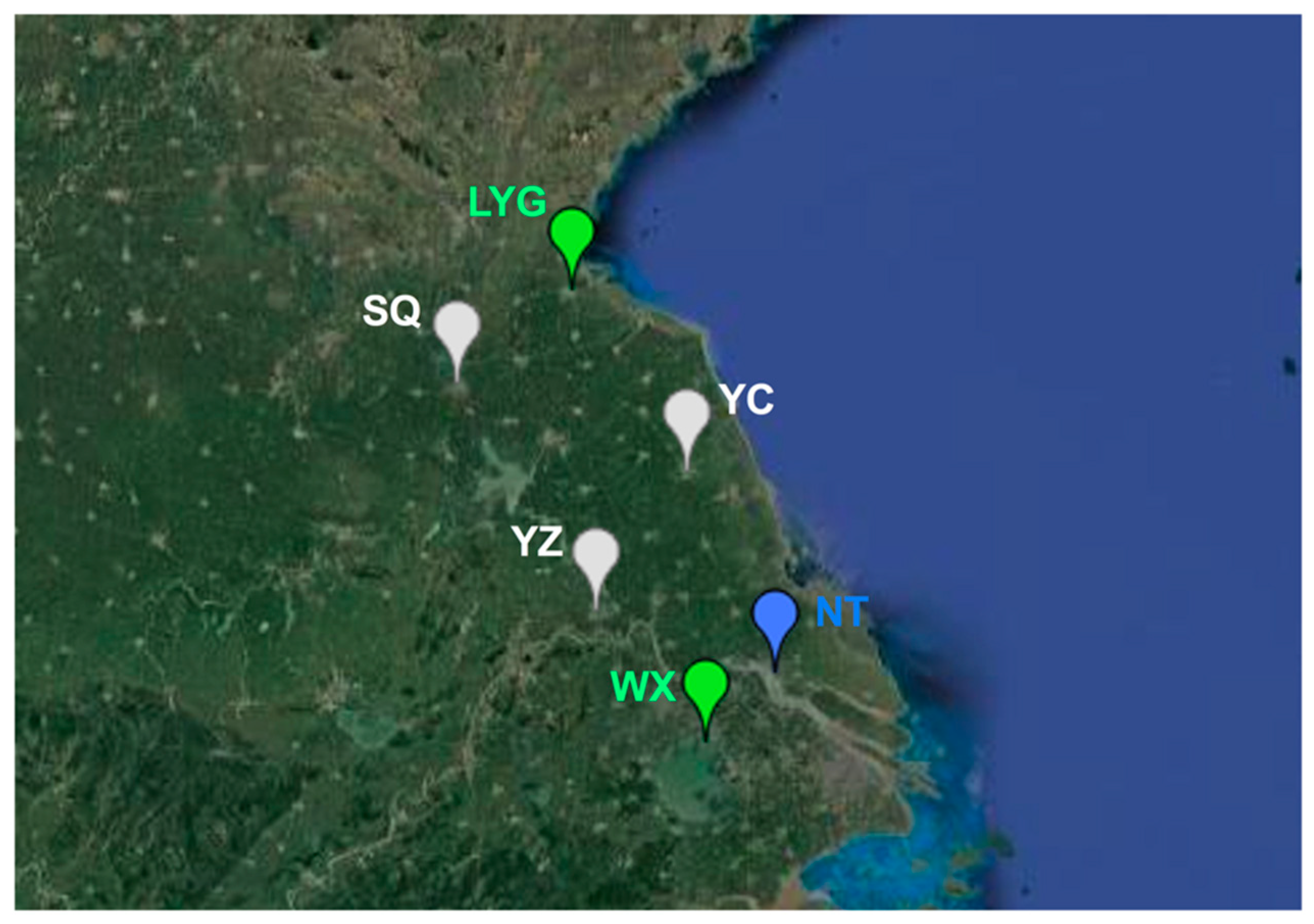
2.1. Sample Collection
2.2. DNA Extraction and Amplification
2.3. Data Analyses
3. Results
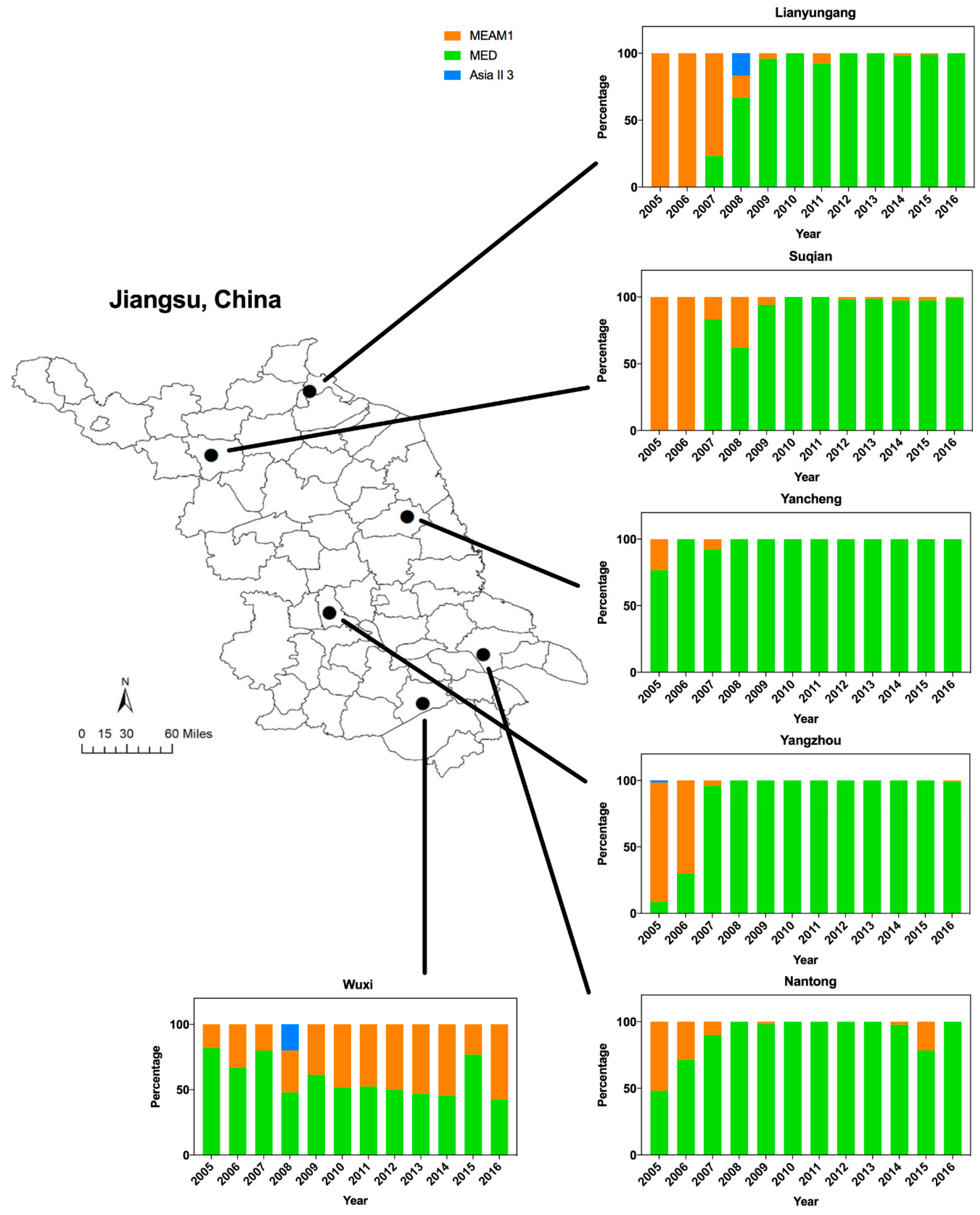
3.1. Dynamics of Whitefly Cryptic Species in Jiangsu from 2005 to 2016
3.2. Genetic Variation
3.3. Population Genetic Structure of MED
3.4. Neutrality Test and Bayesian Skyline Plot
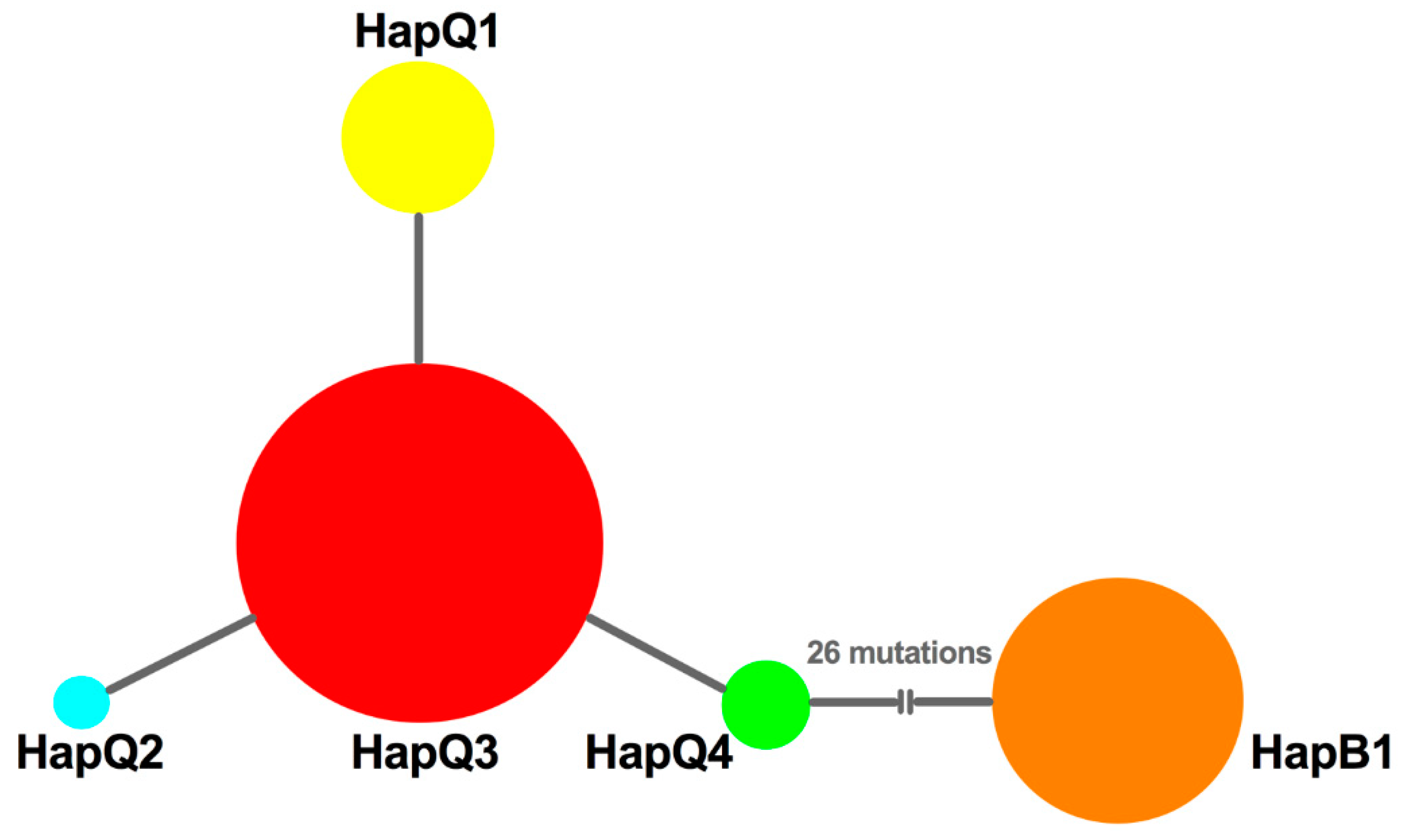
3.5. Phylogenetic Reconstruction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, Y.; Reitz, S.R. Emerging themes in our understanding of species displacements. Annu. Rev. Entomol. 2017, 62, 165–183. [Google Scholar] [CrossRef] [PubMed]
- De Barro, P.J.; Liu, S.-S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, A.; Cook, L.; Riginos, C.; Buckley, Y.; De Barro, P. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Jiang, Z.; Zhang, F.; Liu, Y.; Li, Z.; Zhang, Z. New putative cryptic species detection and genetic network analysis of Bemisia tabaci (Hempitera: Aleyrodidae) in China based on mitochondrial COI sequences. Mitochondrial DNA Part A 2018, 29, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Li, P.; Liu, S.S. Whitefly interactions with plants. Curr. Opin. Insect Sci. 2017, 19, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Boykin, L.M.; Armstrong, K.F.; Kubatko, L.; De Barro, P. Species delimitation and global biosecurity. Evol. Bioinform. 2012, 8, EBO-S8532. [Google Scholar] [CrossRef]
- Chu, D.; Zhang, Y.; Cong, B.; Xu, B.; Wu, Q. Identification for Yunnan Q-biotype Bemisia tabaci population. Entomol. Knowl. 2005, 42, 59–62. [Google Scholar]
- Chu, D.; Zhang, Y.-J.; Brown, J.K.; Cong, B.; Xu, B.-Y.; Wu, Q.-J.; Zhu, G.-R. The introduction of the exotic Q biotype of Bemisia tabaci from the Mediterranean region into China on ornamental crops. Fla. Entomol. 2006, 89, 168–174. [Google Scholar] [CrossRef]
- Teng, X.; Wan, F.-H.; Chu, D. Bemisia tabaci biotype Q dominates other biotypes across China. Fla. Entomol. 2010, 93, 363–368. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, H.; Yang, Y.; Wu, Y. Biotype and insecticide resistance status of the whitefly Bemisia tabaci from China. Pest Manag. Sci. 2010, 66, 1360–1366. [Google Scholar] [CrossRef]
- Hu, J.; De Barro, P.; Zhao, H.; Wang, J.; Nardi, F.; Liu, S.-S. An extensive field survey combined with a phylogenetic analysis reveals rapid and widespread invasion of two alien whiteflies in China. PLoS ONE 2011, 6, e16061. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chu, D.; Ge, D.; Wang, S.; Wu, Q.; Xie, W.; Jiao, X.; Liu, B.; Yang, X.; Yang, N. Further spread of and domination by Bemisia tabaci (Hemiptera: Aleyrodidae) biotype Q on field crops in China. J. Econ. Entomol. 2011, 104, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Luo, C.; Zhang, H.; Guo, X.; Devine, G. Distribution and dynamics of Bemisia tabaci invasive biotypes in central China. Bull. Entomol. Res. 2011, 101, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-S.; De Barro, P.; Xu, J.; Luan, J.-B.; Zang, L.-S.; Ruan, Y.-M.; Wan, F.-H. Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science 2007, 318, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yan, F.; Chu, D.; Pan, H.; Jiao, X.; Xie, W.; Wu, Q.; Wang, S.; Xu, B.; Zhou, X. Difference in feeding behaviors of two invasive whiteflies on host plants with different suitability: Implication for competitive displacement. Int. J. Biol. Sci. 2012, 8, 697. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.-B.; Li, J.; Liu, Y.-Q.; Crowder, D.W.; Liu, S.-S. Effects of reproductive interference on the competitive displacement between two invasive whiteflies. Bull. Entomol. Res. 2014, 104, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Jones, C.; Devine, G.; Zhang, F.; Denholm, I.; Gorman, K. Insecticide resistance in Bemisia tabaci biotype Q (Hemiptera: Aleyrodidae) from China. Crop Prot. 2010, 29, 429–434. [Google Scholar] [CrossRef]
- Sun, D.-B.; Liu, Y.-Q.; Qin, L.; Xu, J.; Li, F.-F.; Liu, S.-S. Competitive displacement between two invasive whiteflies: Insecticide application and host plant effects. Bull. Entomol. Res. 2013, 103, 344–353. [Google Scholar] [CrossRef]
- De Barro, P.; Bourne, A. Ovipositional host choice by an invader accelerates displacement of its indigenous competitor. Biol. Invasions 2010, 12, 3013–3023. [Google Scholar] [CrossRef]
- De Barro, P.; Bourne, A.; Khan, S.; Brancatini, V. Host plant and biotype density interactions–their role in the establishment of the invasive B biotype of Bemisia tabaci. Biol. Invasions 2006, 8, 287–294. [Google Scholar] [CrossRef]
- Chu, D.; Tao, Y.L.; Zhang, Y.J.; Wan, F.H.; Brown, J.K. Effects of host, temperature and relative humidity on competitive displacement of two invasive Bemisia tabaci biotypes [Q and B]. Insect Sci. 2012, 19, 595–603. [Google Scholar] [CrossRef]
- Xiao, N.; Pan, L.-L.; Zhang, C.-R.; Shan, H.-W.; Liu, S.-S. Differential tolerance capacity to unfavourable low and high temperatures between two invasive whiteflies. Sci. Rep. 2016, 6, 24306. [Google Scholar] [CrossRef] [PubMed]
- Jiu, M.; Zhou, X.-P.; Tong, L.; Xu, J.; Yang, X.; Wan, F.-H.; Liu, S.-S. Vector-virus mutualism accelerates population increase of an invasive whitefly. PLoS ONE 2007, 2, e182. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, H.; Jiang, K.; Zhou, X.P.; Liu, S.S. Differential indirect effects of two plant viruses on an invasive and an indigenous whitefly vector: Implications for competitive displacement. Ann. Appl. Biol. 2009, 155, 439–448. [Google Scholar] [CrossRef]
- Li, H.-R.; Pan, H.-P.; Tao, Y.-L.; Zhang, Y.-J.; Chu, D. Population genetics of an alien whitefly in China: Implications for its dispersal and invasion success. Sci. Rep. 2017, 7, 2228. [Google Scholar] [CrossRef] [PubMed]
- Miura, O. Molecular genetic approaches to elucidate the ecological and evolutionary issues associated with biological invasions. Ecol. Res. 2007, 22, 876–883. [Google Scholar] [CrossRef]
- Tsutsui, N.D.; Suarez, A.V.; Holway, D.A.; Case, T.J. Reduced genetic variation and the success of an invasive species. Proc. Natl. Acad. Sci. USA 2000, 97, 5948–5953. [Google Scholar] [CrossRef]
- Crawford, K.; Whitney, K. Population genetic diversity influences colonization success. Mol. Ecol. 2010, 19, 1253–1263. [Google Scholar] [CrossRef]
- Shen, Y.; Du, Y.-Z.; Ren, S.; Qiu, B. Preliminary study of succession of Bemisia tabaci biotypes in Jiangsu Province, China. Chin. J. Appl. Entomol. 2011, 48, 16–21. [Google Scholar]
- Luo, C.; Yao, Y.; Wang, R.; Yan, F.; Hu, D.; Zhang, Z. The use of mitochondrial cytochrome oxidase I (mt CO I) gene sequences for the identification of biotypes of Bemisia tabaci (Gennadius) in China. Acta Entomol. Sin. 2002, 45, 757–763. [Google Scholar]
- Shatters, R.G., Jr.; Powell, C.A.; Boykin, L.M.; Liansheng, H.; McKenzie, C. Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: Development of universal and Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. J. Econ. Entomol. 2009, 102, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Guindon, S.; Lethiec, F.; Duroux, P.; Gascuel, O. PHYML Online—A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, 33, W557–W559. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 10, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [PubMed]
- Ray, N.; Currat, M.; Excoffier, L. Intra-deme molecular diversity in spatially expanding populations. Mol. Biol. Evol. 2003, 20, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L. Patterns of DNA sequence diversity and genetic structure after a range expansion: Lessons from the infinite-island model. Mol. Ecol. 2004, 13, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Vitousek, P.M.; Loope, L.L.; Westbrooks, R. Biological Invasions As Global Environmental Change; USDA: Washington, DC, USA, 1996; Volume 84, pp. 468–478. [Google Scholar]
- Chu, D.; Wan, F.H.; Zhang, Y.J.; Brown, J.K. Change in the biotype composition of Bemisia tabaci in Shandong Province of China from 2005 to 2008. Environ. Entomol. 2010, 39, 1028–1036. [Google Scholar] [CrossRef]
- Chu, D.; Zhang, Y.J.; Wan, F.H. Cryptic invasion of the exotic Bemisia tabaci biotype Q occurred widespread in Shandong Province of China. Fla. Entomol. 2010, 93, 203–207. [Google Scholar] [CrossRef]
- Guo, X.-J.; Rao, Q.; Zhang, F.; Chen, L.; Zhang, H.-Y.; Gao, X.-W. Diversity and genetic differentiation of the whitefly Bemisia tabaci species complex in China based on mtCOI and cDNA-AFLP analysis. J. Integr. Agric. 2012, 11, 206–214. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Zhang, W.; Wu, Q.; Xu, B.; Chu, D. Analysis of genetic diversity among different geographical populations and determination of biotypes of Bemisia tabaci in China. J. Appl. Entomol. 2005, 129, 121–128. [Google Scholar] [CrossRef]
- Chu, D.; Gao, C.; De Barro, P.; Wan, F.; Zhang, Y. Investigation of the genetic diversity of an invasive whitefly (Bemisia tabaci) in China using both mitochondrial and nuclear DNA markers. Bull. Entomol. Res. 2011, 101, 467–475. [Google Scholar] [CrossRef]
- Rollins, L.A.; Moles, A.T.; Lam, S.; Buitenwerf, R.; Buswell, J.M.; Brandenburger, C.R.; Flores-Moreno, H.; Nielsen, K.B.; Couchman, E.; Brown, G.S. High genetic diversity is not essential for successful introduction. Ecol. Evol. 2013, 3, 4501–4517. [Google Scholar] [CrossRef]
- Ren, M.X.; Zhang, Q.G.; Zhang, D.Y. Random amplified polymorphic DNA markers reveal low genetic variation and a single dominant genotype in Eichhornia crassipes populations throughout China. Weed Res. 2005, 45, 236–244. [Google Scholar] [CrossRef]
- Zimmermann, H.; Ritz, C.M.; Hirsch, H.; Renison, D.; Wesche, K.; Hensen, I. Highly reduced genetic diversity of Rosa rubiginosa L. populations in the invasive range. Int. J. Plant Sci. 2010, 171, 435–446. [Google Scholar] [CrossRef]
- Frankham, R. Resolving the genetic paradox in invasive species. Heredity 2005, 94, 385. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, J.J.; Glor, R.E.; Schettino, L.R.; Lara, A.C.; Larson, A.; Losos, J.B. Genetic variation increases during biological invasion by a Cuban lizard. Nature 2004, 431, 177. [Google Scholar] [CrossRef] [PubMed]
- Genton, B.; Shykoff, J.; Giraud, T. High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol. Ecol. 2005, 14, 4275–4285. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, P.; Zhang, Y.-P.; Luo, J.; Lundeberg, J.; Leitner, T. Genetic evidence for an East Asian origin of domestic dogs. Science 2002, 298, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, P.; Jiang, F.; CHAPUIS, M.P.; Shali, Y.; Sword, G.A.; Kang, L. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef]
- Zhang, B.; Edwards, O.; Kang, L.; Fuller, S. Russian wheat aphids (Diuraphis noxia) in China: Native range expansion or recent introduction? Mol. Ecol. 2012, 21, 2130–2144. [Google Scholar] [CrossRef]
- Bertelsmeier, C.; Ollier, S.; Liebhold, A.M.; Brockerhoff, E.G.; Ward, D.; Keller, L. Recurrent bridgehead effects accelerate global alien ant spread. Proc. Natl. Acad. Sci. USA 2018, 115, 5486–5491. [Google Scholar] [CrossRef]
- Chu, D.; Wan, F.H.; Tao, Y.L.; Liu, G.X.; Fan, Z.X.; Bi, Y.P. Genetic differentiation of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) biotype Q based on mitochondrial DNA markers. Insect Sci. 2008, 15, 115–123. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Index | Lianyungang | Suqian | Yancheng | Yangzhou | Nantong | Wuxi |
|---|---|---|---|---|---|---|
| Haplotype Diversity (H) | 0.080 | 0.220 | 0.285 | 0.215 | 0.352 | 0.000 |
| Nucleotide Diversity (π) | 0.00014 | 0.00039 | 0.00039 | 0.00039 | 0.00078 | 0.00000 |
| Populations | Lianyungang | Suqian | Yancheng | Yangzhou | Nantong | Wuxi |
|---|---|---|---|---|---|---|
| Lianyungang | − | − | − | − | − | |
| Suqian | 0.02640 | − | − | − | − | |
| Yancheng | 0.03546 | −0.03945 | − | − | + | |
| Yangzhou | 0.01216 | −0.02506 | −0.00666 | − | − | |
| Nantong | 0.02413 | 0.01178 | 0.03126 | 0.02137 | + | |
| Wuxi | 0.00370 | 0.13861 | 0.10941 | 0.04131 | 0.05545 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.-T.; Cai, L.; Shen, Y.; Xu, L.-L.; Du, Y.-Z. Competitive Displacement between Bemisia tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED. Insects 2020, 11, 35. https://doi.org/10.3390/insects11010035
Tang X-T, Cai L, Shen Y, Xu L-L, Du Y-Z. Competitive Displacement between Bemisia tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED. Insects. 2020; 11(1):35. https://doi.org/10.3390/insects11010035
Chicago/Turabian StyleTang, Xiao-Tian, Li Cai, Yuan Shen, Li-Li Xu, and Yu-Zhou Du. 2020. "Competitive Displacement between Bemisia tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED" Insects 11, no. 1: 35. https://doi.org/10.3390/insects11010035
APA StyleTang, X.-T., Cai, L., Shen, Y., Xu, L.-L., & Du, Y.-Z. (2020). Competitive Displacement between Bemisia tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED. Insects, 11(1), 35. https://doi.org/10.3390/insects11010035