Population Genetics of Bactrocera minax (Diptera: Tephritidae) in China Based on nad4 Gene Sequence


 and
and
Abstract
1. Introduction
2. Materials and Methods
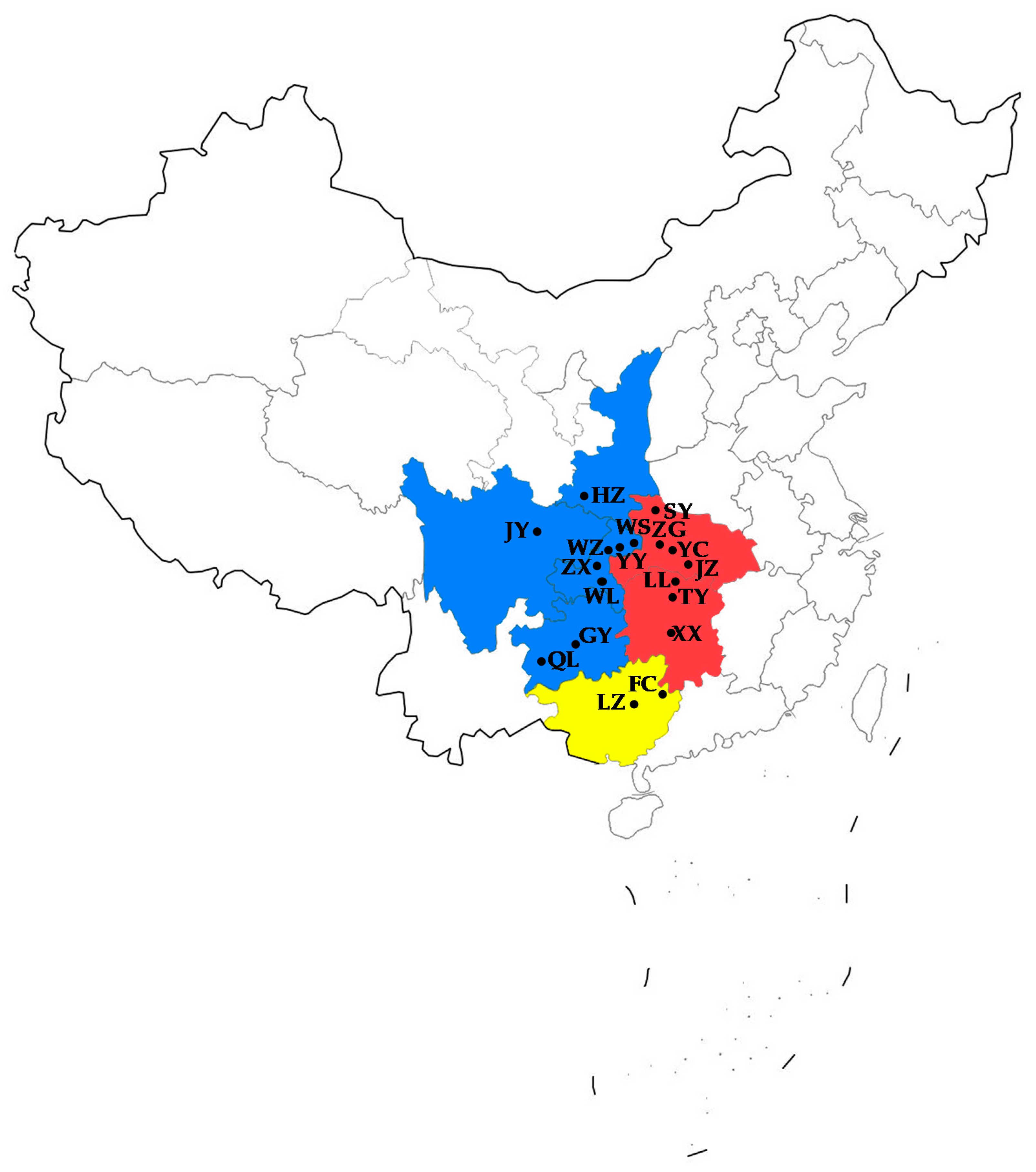
2.1. Sampling
2.2. DNA Extraction and Amplification
2.3. Data Analysis
3. Results
3.1. Genetic Diversity
3.2. Population Genetic Structure
3.3. Estimation of Migration Rates
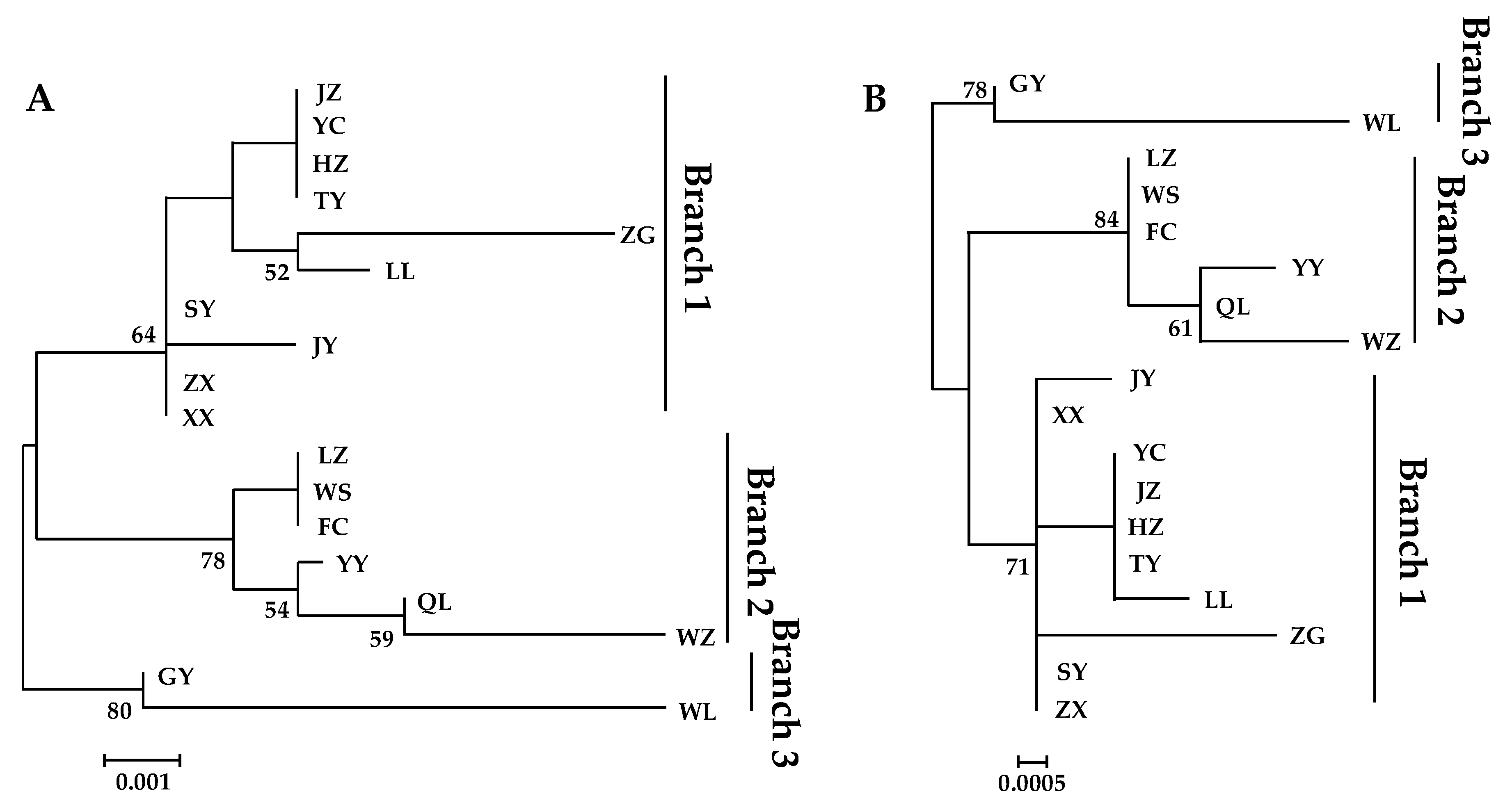
3.4. Phylogenetic Analysis
3.4.1. Phylogenetic Trees Construction
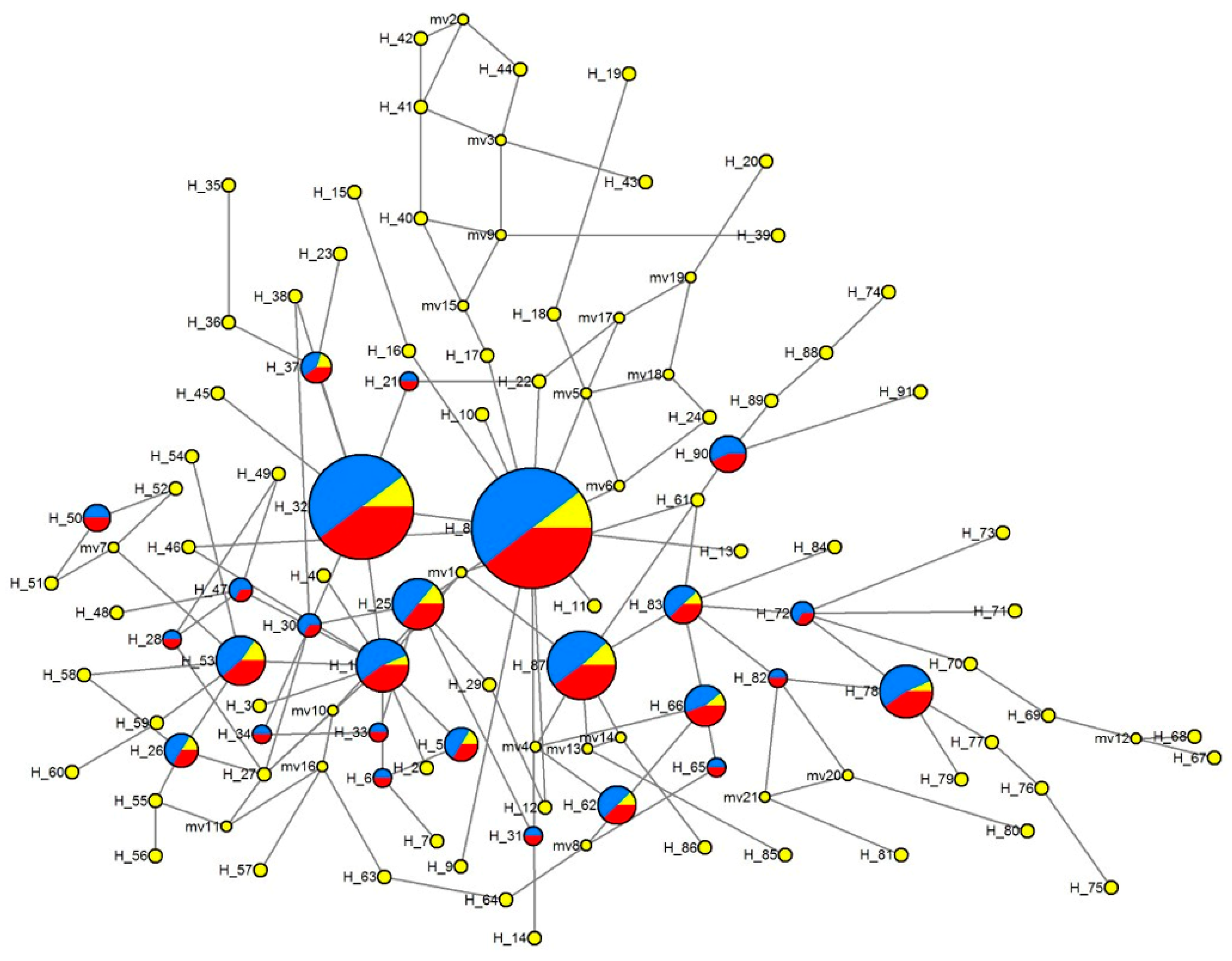
3.4.2. Median-Joining Network of Haplotypes
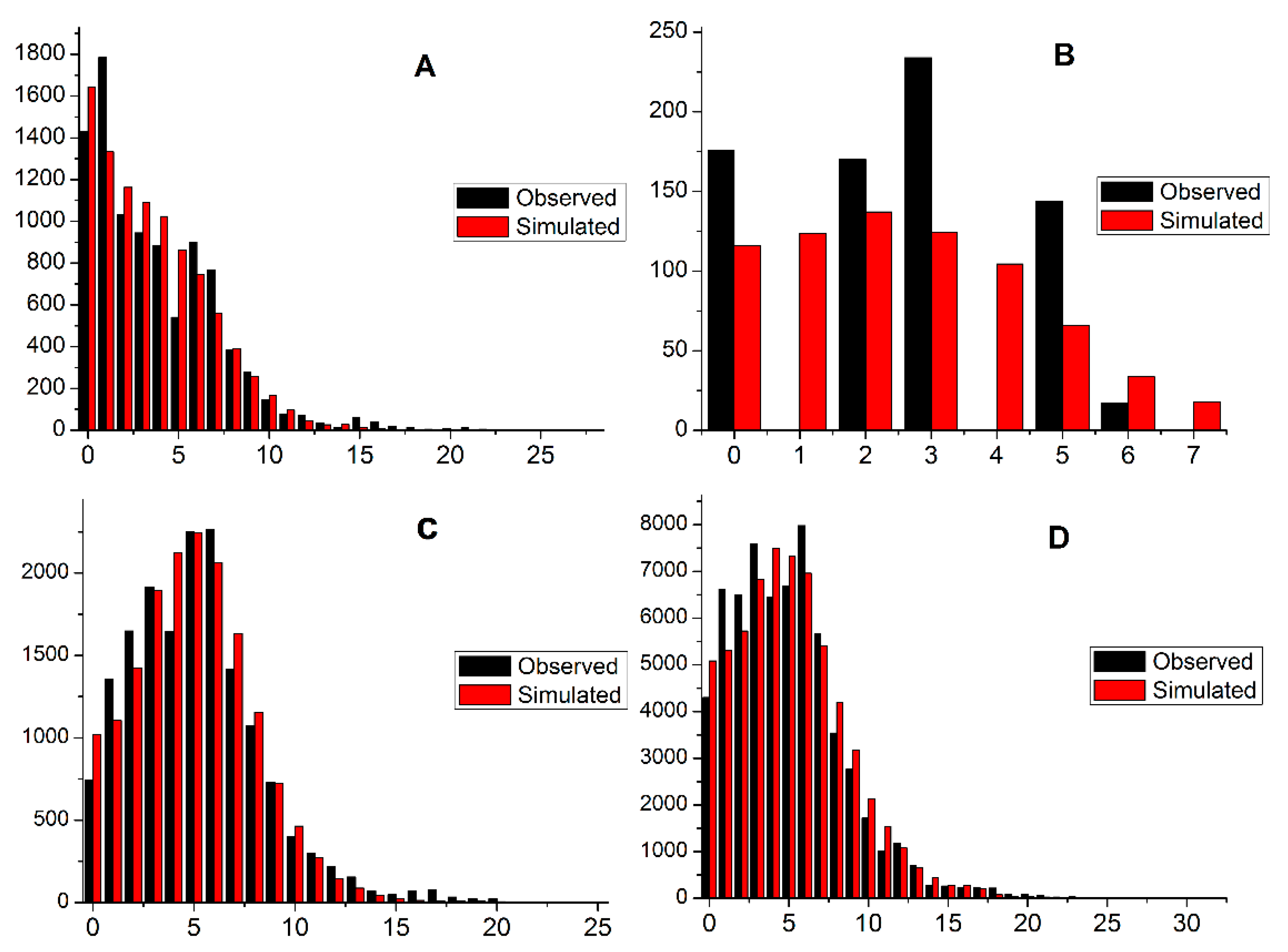
3.5. Demographic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, E.H.; Dou, W.; Hu, F.; Tang, S.; Zhao, Z.M.; Wang, J.J. Purification and biochemical characterization of glutathione S-transferases in Bactrocera minax (Diptera: Tephritidae). Fla. Entomol. 2012, 95, 593–601. [Google Scholar] [CrossRef]
- Drew, R.A.I.; Dorji, C.; Romig, M.C.; Loday, P. Attractiveness of various combinations of colors and shapes to females and males of Bactrocera minax (Diptera: Tephritidae) in a commercial mandarin grove in Bhutan. J. Econ. Entomol. 2006, 99, 1651–1656. [Google Scholar] [CrossRef]
- Gao, L.Z.; Liu, Y.H.; Wan, X.W.; Wang, J.; Hong, F. Screening of microsatellite markers in Bactrocera minax (Diptera: Tephritidae). Sci. Agric. Sin. 2013, 46, 3285–3292. [Google Scholar]
- Chen, S.X.; Xie, Y.Z. Taxonomic notes on the Chinese citrus fly Tetradacus citri (Chen). Acta Entomol. Sin. 1955, 1, 123–126. [Google Scholar]
- Dorji, C.; Clarke, A.R.; Drew, R.A.I.; Fletcher, B.S.; Loday, P.; Mahat, K.; Raghu, S.; Romig, M.C. Seasonal phenology of Bactrocera minax (Diptera: Tephritidae) in western Bhutan. Bull. Entomol. Res. 2006, 96, 531–538. [Google Scholar]
- Zhou, X.W.; Niu, C.Y.; Han, P.; Desneux, N. Field evaluation of attractive lures for the fruit fly Bactrocera minax (Diptera: Tephritidae) and their potential use in spot sprays in Hubei Province (China). J. Econ. Entomol. 2012, 105, 1277–1284. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Y.H.; Wan, X.W.; Wang, Z.L. Molecular phylogeny of Bactrocera species (Diptera: Tephritidae: Dacini) inferred from mitochondrial sequences of 16S rDNA and COI sequences. Fla. Entomol. 2010, 93, 369–377. [Google Scholar]
- Zhang, C.Y. Molecular Phylogenetic Study and Molecular Identification in Five Species of Fruit Flies (Diptera:Tephritidae). Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, June 2007. [Google Scholar]
- Wan, X.W.; Liu, Y.H.; Zhang, B. Invasion history of the oriental fruit fly, Bactrocera dorsalis, in the Pacific-Asia region: Two main invasion routes. PLoS ONE 2012, 7, e36176. [Google Scholar] [CrossRef]
- Wan, X.; Nardi, F.; Zhang, B.; Liu, Y. The oriental fruit fly, Bactrocera dorsalis, in China: Origin and gradual inland range expansion associated with population growth. PLoS ONE 2011, 6, e25238. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Liu, J.; Berry, R.E.; Blouin, M.S. Molecular differentiation and phylogeny of entomopathogenic nematodes (Rhabditida: Heterorhabditidae) based on ND4 gene sequences of mitochondrial DNA. J. Parasitol. 1999, 85, 709–715. [Google Scholar] [CrossRef]
- Zhang, B.; Nardi, F.; Hull-Sanders, H.; Wan, X.; Liu, Y. The complete nucleotide sequence of the mitochondrial genome of Bactrocera minax (Diptera: Tephritidae). PLoS ONE 2014, 9, e100558. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Polzin, T.; Daneshmand, S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Zhang, B. Analysis of Complete Mitochondrial Genome of Bactrocera minax (Diptera: Tephritidae) and Exploration of Identification and Detection for Dangerous Tephritid Species by Microarray. Ph.D. Thesis, Southwest University, Chongqing, China, December 2010. [Google Scholar]
- Brower, A.V.Z. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial-DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef]
- Allendorf, F.W. Isolation, gene flow, and genetic differentiation among populations. In Genetics and Conservation; Schonewald-Cox, C.M., Chambers, S.M., MacBryde, B., Thomas, W.L., Eds.; Benjamin/Cummings: Menlo Park, CA, USA, 1983; pp. 51–65. [Google Scholar]
- Bellemain, E.; Gaggiotti, O.E.; Fahey, A.; Bermingham, E.; Ricklefs, R.E. Demographic history and genetic diversity in West Indian Coereba flaveola populations. Genetica 2012, 140, 137–148. [Google Scholar] [CrossRef]
- Brooker, L.; Brooker, M. Dispersal and population dynamics of the blue-breasted fairy-wren, Malurus pulcherrimus, in fragmented habitat in the Western Australian wheatbelt. Wildl. Res. 2002, 29, 225–233. [Google Scholar] [CrossRef]
- Gibbs, J.P. Demography versus habitat fragmentation as determinants of genetic variation in wild populations. Biol. Conserv. 2001, 100, 15–20. [Google Scholar] [CrossRef]
- Templeton, A.R.; Robertson, R.J.; Brisson, J.; Strasburg, J. Disrupting evolutionary processes: The effect of habitat fragmentation on collared lizards in the Missouri Ozarks. Proc. Natl. Acad. Sci. USA 2001, 98, 5426–5432. [Google Scholar] [CrossRef]
- Whitlock, M.C.; Barton, N.H. The effective size of a subdivided population. Genetics 1997, 146, 427–441. [Google Scholar]
- Johnson, J.A.; Toepfer, J.E.; Dunn, P.O. Contrasting patterns of mitochondrial and microsatellite population structure in fragmented populations of greater prairie-chickens. Mol. Ecol. 2003, 12, 3335–3347. [Google Scholar] [CrossRef]
- Gao, L.; Liu, Y.; Wan, X.; Tu, Z.; Pu, P.; Chen, Y. Genetic diversity analysis of Bactrocera minax (Diptera: Tephritidae) in China. Plant Prot. 2016, 42, 51–55. [Google Scholar]
- Bachtrog, D.; Andolfatto, P. Selection, recombination and demographic history in Drosophila miranda. Genetics 2006, 174, 2045–2059. [Google Scholar] [CrossRef]
- Haddrill, P.R.; Thornton, K.R.; Charlesworth, B.; Andolfatto, P. Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 2005, 15, 790–799. [Google Scholar] [CrossRef][Green Version]
- Cavallis, L.L.; Edwards, A.W.F. Phylogenetic analysis: Models and estimation procedures. Evolution 1967, 21, 550–570. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location (City, Province) | Abbreviation | Number of Individuals | Longitude | Latitude |
|---|---|---|---|---|
| Shiyan, Hubei | SY | 21 | 110°28′(E) | 32°23′(N) |
| Zigui, Hubei | ZG | 21 | 110°58′(E) | 30°49′(N) |
| Yichang, Hubei | YC | 19 | 111°19′(E) | 30°46′(N) |
| Jingzhou, Hubei | JZ | 20 | 112°14′(E) | 30°20′(N) |
| Xiangxi, Hunan | XX | 16 | 109°44′(E) | 28°18′(N) |
| Taoyuan, Hunan | TY | 21 | 111°28′(E) | 28°54′(N) |
| Linli, Hunan | LL | 20 | 111°38′(E) | 29°26′(N) |
| Fuchuan, Guangxi | FC | 19 | 111°16′(E) | 24°38′(N) |
| Luzhai, Guangxi | LZ | 20 | 109°43′(E) | 24°28′(N) |
| Qinglong, Guizhou | QL | 20 | 105°12′(E) | 25°50′(N) |
| Guiyang, Guizhou | GY | 21 | 106°39′(E) | 26°24′(N) |
| Wulong, Chongqing | WL | 18 | 107°45′(E) | 29°19′(N) |
| Zhongxian, Chongqing | ZX | 21 | 108°01′(E) | 30°18′(N) |
| Wanzhou, Chongqing | WZ | 19 | 108°24′(E) | 30°48′(N) |
| Yunyang, Chongqing | YY | 20 | 108°41′(E) | 30°55′(N) |
| Wushan, Chongqing | WS | 23 | 109°52′(E) | 31°04′(N) |
| Hanzhong, Shanxi | HZ | 20 | 107°01′(E) | 33°04′(N) |
| Jiangyou, Sichuan | JY | 20 | 104°44′(E) | 31°46′(N) |
| Population | S | h | k | π | Tajima’s D | Fu’s FS | τ | PSSD |
|---|---|---|---|---|---|---|---|---|
| SY | 4 | 0.724 | 1.095 | 0.002 | 0.787 | −27.046 ** | 3.742 | 0.180 |
| ZG | 14 | 0.924 | 3.824 | 0.007 | 0.157 | −24.443 ** | 4.926 | 0.390 |
| YC | 7 | 0.526 | 1.076 | 0.002 | −1.558 * | −20.489 ** | 10.309 | 0.480 |
| JZ | 5 | 0.432 | 0.837 | 0.001 | −1.243 | −34.028 ** | 0.004 | 0.000 |
| XX | 5 | 0.425 | 1.225 | 0.002 | −0.274 | −28.882 ** | 0.066 | 0.000 |
| TY | 15 | 0.810 | 2.457 | 0.004 | −1.535 * | −26.755 ** | 0.525 | 0.000 |
| LL | 23 | 0.800 | 3.105 | 0.005 | −1.778 * | −23.195 ** | 0.352 | 0.000 |
| FC | 6 | 0.708 | 2.117 | 0.003 | 0.755 | −26.845 ** | 3.414 | 0.040 |
| LZ | 9 | 0.795 | 2.937 | 0.005 | 0.542 | −25.571 ** | 4.621 | 0.040 |
| QL | 14 | 0.837 | 3.474 | 0.006 | −0.849 | −21.614 ** | 6.227 | 0.120 |
| GY | 11 | 0.886 | 2.405 | 0.004 | −0.355 | −25.177 ** | 3.818 | 0.170 |
| WL | 21 | 0.928 | 5.327 | 0.009 | −0.499 | −15.610 ** | 5.787 | 0.410 |
| ZX | 0 | 0.000 | 0.000 | 0.000 | 0.000 | 34.028 | 0.000 | 0.000 |
| WZ | 28 | 0.386 | 3.146 | 0.005 | −2.398 ** | −23.178 ** | 3.000 | 0.160 |
| YY | 16 | 0.868 | 2.832 | 0.005 | −1.383 | −25.864 ** | 2.236 | 0.670 |
| WS | 11 | 0.818 | 2.996 | 0.005 | 0.018 | −26.357 ** | 4.125 | 0.340 |
| HZ | 2 | 0.479 | 0.958 | 0.002 | 1.639 | −34.028 ** | 2.761 | 0.160 |
| JY | 22 | 0.837 | 5.584 | 0.010 | −0.215 | −17.651 ** | 6.268 | 0.090 |
| All | 93 | 0.917 | 4.217 | 0.007 | −1.979 ** | −24.928 ** | 4.287 | 0.860 |
| CC | −1.923 ** | −25.881 ** | 7.143 | 0.910 | ||||
| SC | 0.534 | −26.622 ** | 3.639 | 0.080 | ||||
| WC | −1.764 ** | −25.194 ** | 4.504 | 0.760 |
| Locus Analyzed | Source of Variation | df. | Sum of Squares | Variance Components | Percentage of Variation | Fixation Indices |
|---|---|---|---|---|---|---|
| nad4 | Among groups | 2 | 202.397 | 1.740 Va | 46.15 | FCT = 0.462 ** |
| Among populations within groups | 15 | 216.748 | 0.597 Vb | 15.84 | FSC = 0.294 ** | |
| Within populations | 341 | 2588.323 | 1.433 Vc | 38.01 | FST = 0.620 ** |
| Group | CC | SC | WC |
|---|---|---|---|
| CC | — | ||
| SC | 0.315 ** | — | |
| WC | 0.211 ** | 0.053 ** | — |
| Group | θ | M | |||
|---|---|---|---|---|---|
| CC→i | SC→i | WC→i | |||
| nad4 | CC | 0.032 (0.013–0.061) | — | 382.3 (64.0–794.7) | 158.3 (0.0–589.3) |
| SC | 0.004 (0.000–0.010) | 298.3 (0.0–825.3) | — | 373.0 (60.0–798.0) | |
| WC | 0.072 (0.034–0.100) | 137.7 (0.0–260.0) | 462.3 (119.3–878.0) | — | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, F.; Gao, L.; Han, H.-L.; Wang, P.; Wang, J.; Wei, D.; Liu, Y. Population Genetics of Bactrocera minax (Diptera: Tephritidae) in China Based on nad4 Gene Sequence. Insects 2019, 10, 236. https://doi.org/10.3390/insects10080236
Hong F, Gao L, Han H-L, Wang P, Wang J, Wei D, Liu Y. Population Genetics of Bactrocera minax (Diptera: Tephritidae) in China Based on nad4 Gene Sequence. Insects. 2019; 10(8):236. https://doi.org/10.3390/insects10080236
Chicago/Turabian StyleHong, Feng, Lizhi Gao, Hong-Liang Han, Pan Wang, Jia Wang, Dong Wei, and Yinghong Liu. 2019. "Population Genetics of Bactrocera minax (Diptera: Tephritidae) in China Based on nad4 Gene Sequence" Insects 10, no. 8: 236. https://doi.org/10.3390/insects10080236
APA StyleHong, F., Gao, L., Han, H.-L., Wang, P., Wang, J., Wei, D., & Liu, Y. (2019). Population Genetics of Bactrocera minax (Diptera: Tephritidae) in China Based on nad4 Gene Sequence. Insects, 10(8), 236. https://doi.org/10.3390/insects10080236