Precision Oncology—The Quest for Evidence


Abstract
1. Introduction
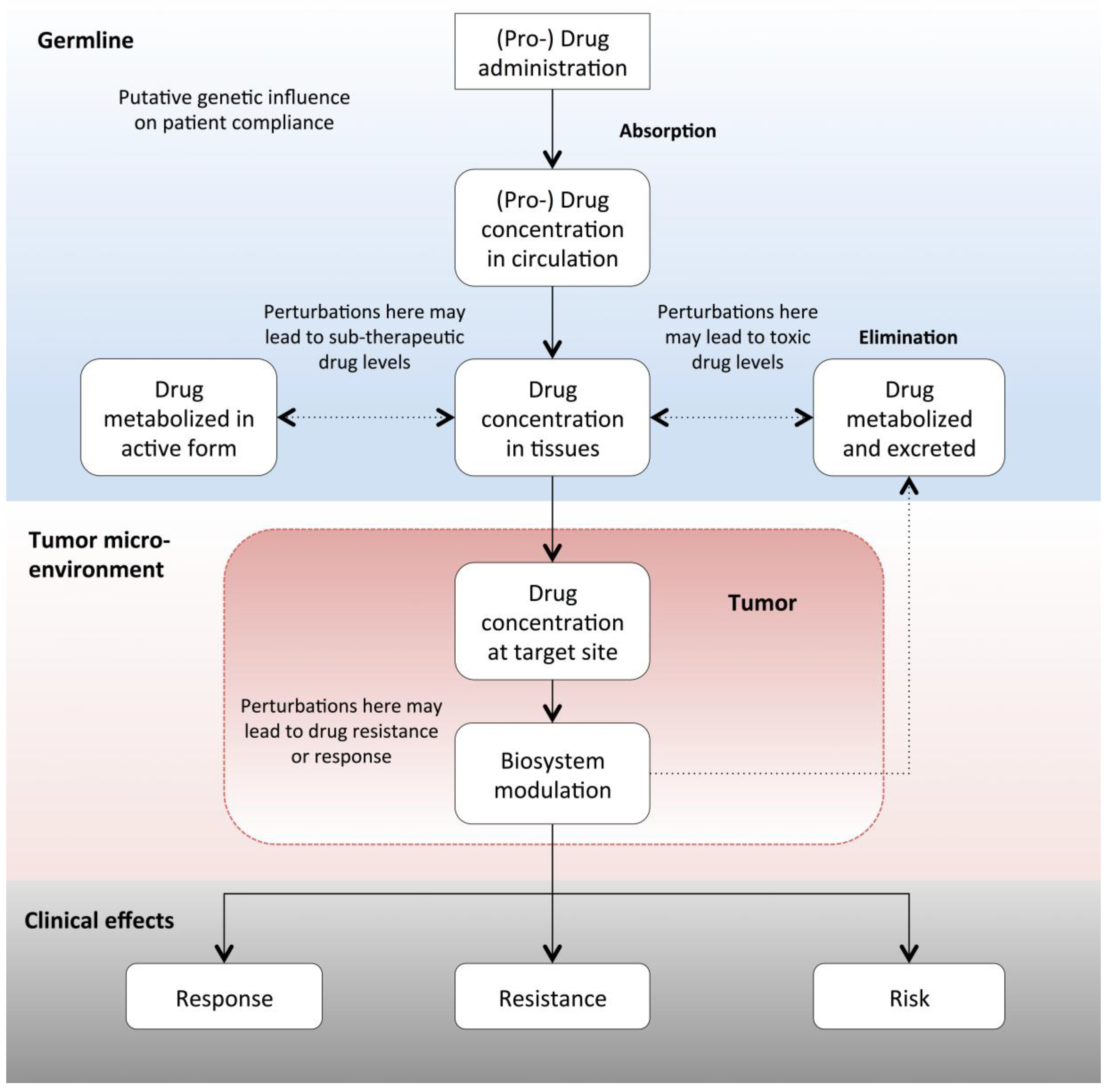
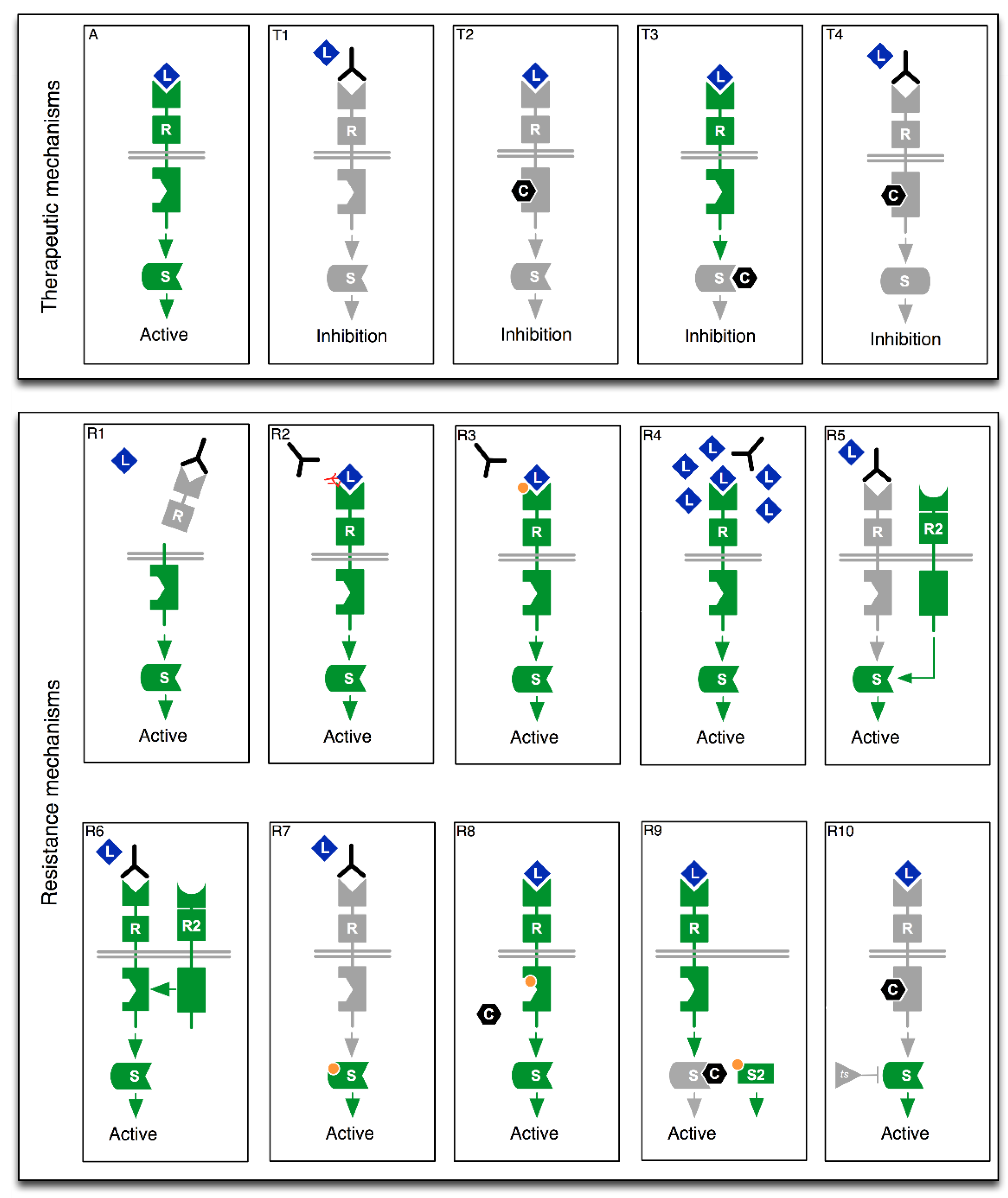
2. From Drug Mode of Action to Precision Oncology
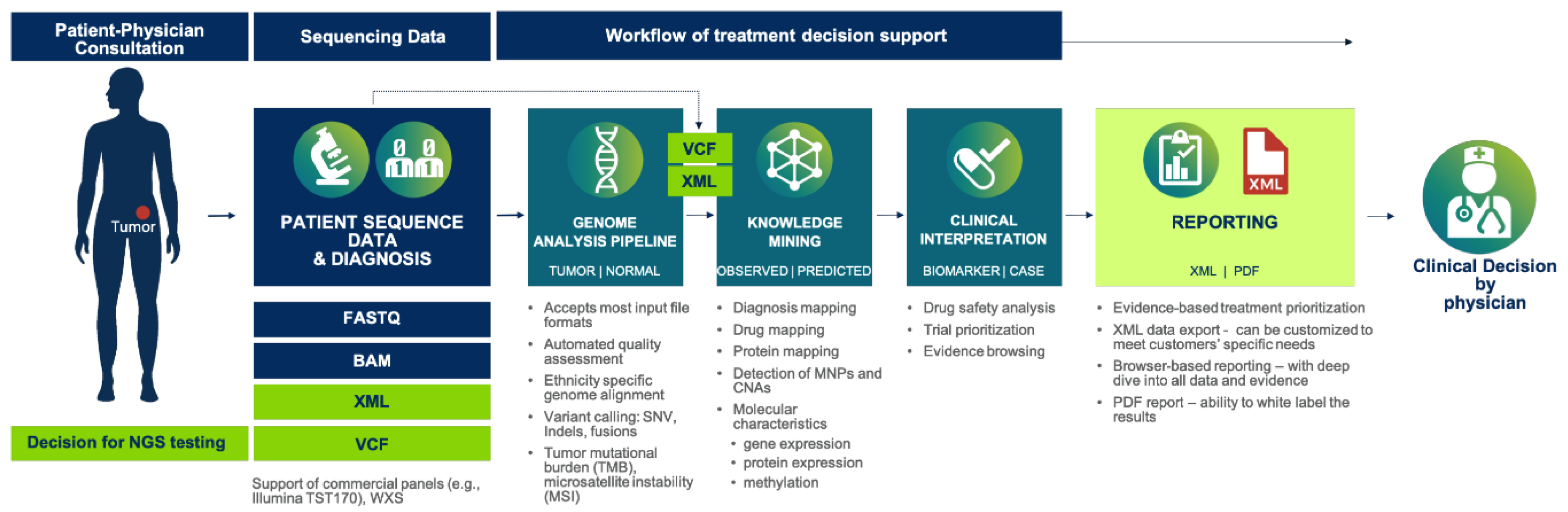
2.1. The Case for Multi-Gene Diagnostic Testing
2.2. Cancer Gene Panels and Analytical Software
- (a)
- Detailed variant descriptions—e.g., the type of genomic aberration (such as SNP, Insertion, or Deletion, etc.)
- (b)
- The relevant drug or treatment
- (c)
- The effect of variant on treatment responsiveness—i.e., response, resistance or toxicity
- (d)
- The quantity of effect—e.g., strong, medium, weak
- (e)
- The observation context (i.e., the disease, disease stage, or model system)
- (f)
- A link to the source information and a grading of its reliability
3. Precision Oncology Trials—The Quest for Evidence
- What is the strength of the clinical evidence that the technology is safe and effective?
- What group of patients, if any, would benefit most from using a given technology for preventing, diagnosing, or treating a particular condition?
- Under what circumstances and conditions, if any, would the technology be most appropriately used?
- How does the new technology compare to other available treatments for the same condition?
3.1. Clinical Trial Strategies
- “Adaptive trials” evolve dynamically based on emergent trial data. This leads to hypotheses optimization and testing where randomization ratios can be modified, treatment arms with inferior outcomes eliminated, and increased biomarker-based assignment results in a higher proportion of patients to be randomly assigned to the more effective treatment arms [24].
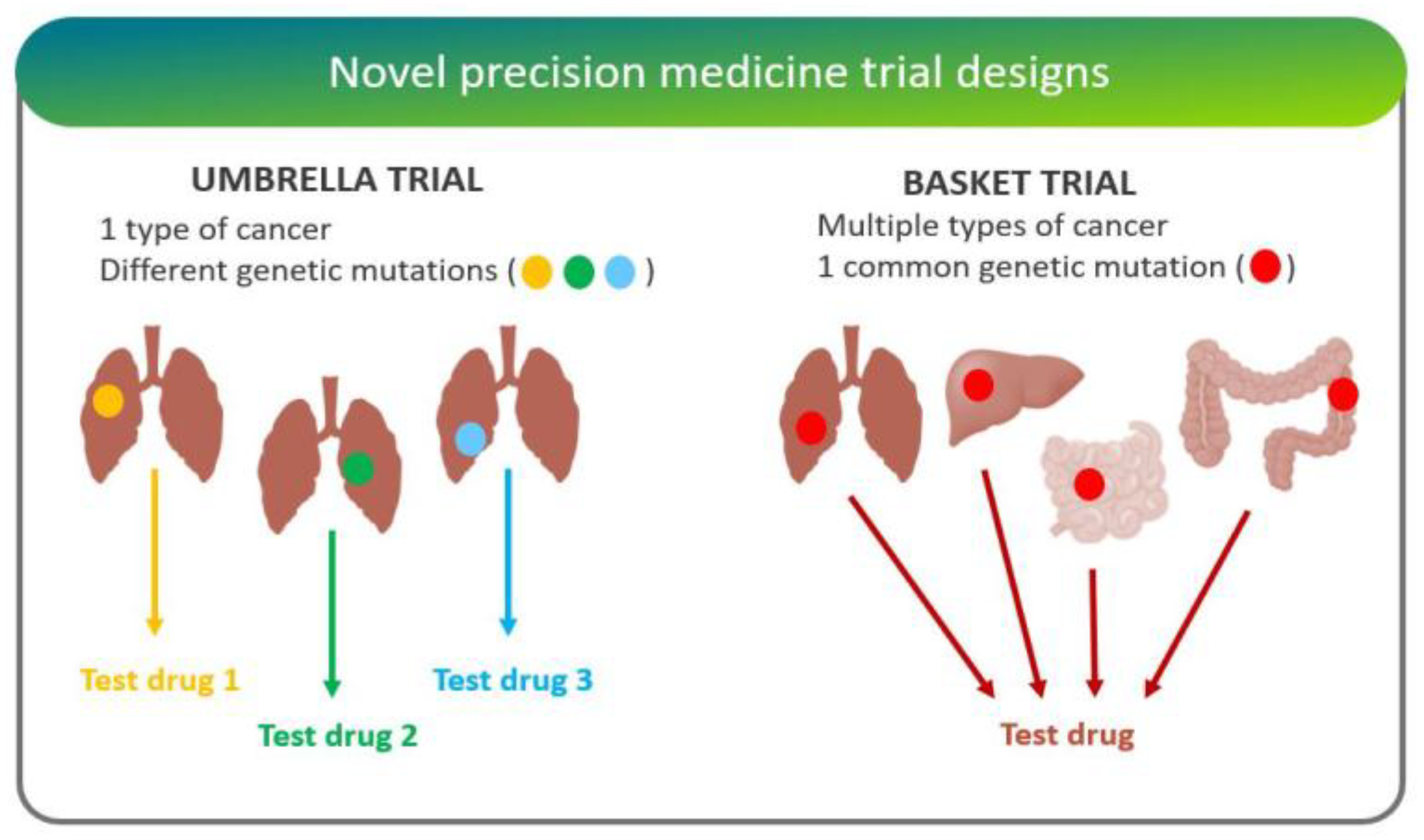
- “Basket trials” test whether a drug is effective in patients with specific genetic alterations regardless of their disease of origin [25]. For instance, the National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH) is a Phase II clinical trial in which patients who share a common genetic mutation for a given cancer are sorted into “baskets”, or treatment arms, regardless of cancer type.
- “Umbrella trials” assign patients to one of potentially many treatment arms, based on a specific cancer type and genetic markers. For example, the Lung Master Protocol (Lung-MAP) study aims to rapidly identify drug therapies for particular cancer types, making it a useful design for cancers with wide genetic heterogeneity [24].
- In “n-of-one trials”, the patient serves as both control and experimental “arm”. This design is especially useful for low-frequency molecular aberrations/conditions, where randomized studies are difficult [26].
- Randomized/Histology-agnostic
- Randomized/Histology-specific
- Non-randomized/Histology-agnostic
- Non-randomized/Histology-specific
3.2. Randomized Histology-Agnostic Studies
3.2.1. IMPACT II (MD Anderson Cancer Center/Foundation Medicine)
3.2.2. M-PACT (NCI)
3.3. Randomized Histology-Specific Studies
3.3.1. BATTLE2 (MD Anderson Cancer Center)
3.3.2. ALCHEMIST (NCI)
3.3.3. LungMAP (SWOG1400)
3.3.4. SAFIR-02 (Lung)
3.3.5. SAFIR-02 (Breast)
3.4. Non-Randomized Histology-Agnostic Studies
3.4.1. MATCH (NCI)
3.4.2. WINtherapeutics
3.4.3. MOSCATO
3.4.4. TAPUR
3.5. Non-Randomized Histology-Specific Studies
3.5.1. Pre-SAFIR
3.5.2. SAFIR-01
3.6. Perspectives
- Number of genes characterized
- Extent of gene characterization (sub-exome, exome, or whole genome)
- Ability to computationally interpret the clinical implications of patients’ clinical and molecular data
- Clinical experience of the treating physician(s)
- Availability of prioritized therapies
4. Discussion
- Which drugs stand the highest likelihood of working in my patient?
- Which drugs stand the highest likelihood of not working in my patient?
- Which drug combinations might be particularly efficacious?
- Which drugs and drug combinations should be contraindicated?
- What dose should a particular drug/combination be given at?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Food & Drug Administration (FDA). Table of Pharmacogenomic Biomarkers in Drug Labeling. 2019. Available online: http://www.fda.gov/drugs/science-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 7 August 2019).
- Food & Drug Administration (FDA). Table of Pharmacogenomic Biomarkers in Drug Labeling. 2018. Available online: https://www.fda.gov/media/124784/download (accessed on 7 August 2019).
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.; Skora, A.; Azad, N.S.; Laheru, D.A.; Donehower, R.C.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Lassen, U.N. TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Target. Oncol. 2018, 13, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Bertucci, F.; Sabatier, R.; Gonçalves, A. Targeting BRCA Deficiency in Breast Cancer: What are the Clinical Evidences and the Next Perspectives? Cancers (Basel) 2018, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Klein, P.; et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matías-Guiu, X.; Cajal, S.R.Y.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- Nagy, P.; Friedländer, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar]
- Jacobs, B.; De Roock, W.; Piessevaux, H.; Van Oirbeek, R.; Biesmans, B.; De Schutter, J.; Fieuws, S.; Vandesompele, J.; Peeters, M.; Van Laethem, J.-L.; et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 2009, 27, 5068–5074. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Nahta, R.; Yuan, L.X.H.; Zhang, B.; Kobayashi, R.; Esteva, F.J. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65, 11118–11128. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.N.; You, F.; Schnitt, S.J.; Witkiewicz, A.; Lu, X.; Sgroi, D.; Ryan, P.D.; Come, S.E.; Burstein, H.J.; Lesnikoski, B.-A.; et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin. Cancer Res. 2007, 13, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Caponigro, G. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef]
- Li, F.-H.; Shen, L.; Li, Z.-H.; Luo, H.-Y.; Qiu, M.-Z.; Zhang, H.-Z.; Li, Y.-H.; Xu, R.-H. Impact of KRAS mutation and PTEN expression on cetuximab-treated colorectal cancer. World J. Gastroenterol. WJG 2010, 16, 5881–5888. [Google Scholar] [CrossRef]
- Shah, P.; Kendall, F.; Khozin, S.; Goosen, R.; Hu, J.; Laramie, J.; Ringel, M.; Schork, N. Artificial intelligence and machine learning in clinical development: A translational perspective. NPJ Digit. Med. 2019, 2, 69. [Google Scholar] [CrossRef]
- Barna, A.; Cruz-Sanchez, T.M.; Brigham, K.B.; Thuong, C.-T.; Kristensen, F.B.; Durand-Zaleski, I. Evidence Required by Health Technolgy Assessment and Reimbursement Bodies Evaluating Diagnostic or Prognostic Algorithms That Include Omics Data. Int. J. Technol. Assess. Health Care 2018, 34, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J.; LaVange, L.M. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N. Engl. J. Med. 2017, 377, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Janiaud, P.; Serghiou, S.; Ioannidis, J.P.A. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat. Rev. 2019, 73, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Billingham, L.; Malottki, K.; Steven, N. Research methods to change clinical practice for patients with rare cancers. Lancet Oncol. 2016, 17, e70–e80. [Google Scholar] [CrossRef]
- West, H.J. Novel Precision Medicine Trial Designs: Umbrellas and Baskets. JAMA Oncol. 2017, 3, 423. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Lee, J.J.; Wistuba, I.I.; Tsao, A.S.; Fossella, F.V.; Kalhor, N.; Gupta, S.; Byers, L.A.; Izzo, J.G.; Gettinger, S.N.; et al. The BATTLE-2 Study: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients with Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3638–3647. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Giaccone, G. Lessons learned from BATTLE-2 in the war on cancer: The use of Bayesian method in clinical trial design. Ann. Transl. Med. 2016, 4, 23. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Stephenson, J.J.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Hong, D.S.; Ye, Y.; Cartwright, C.; Wheler, J.J.; Falchook, G.S.; Naing, A.; Fu, S.; Piha-Paul, S.; Meric-Bernstam, F.; et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis. Oncol. 2017, 1, 1–18. [Google Scholar]
- Tsimberidou, A.-M. Initiative for Molecular Profiling and Advanced Cancer Therapy and challenges in the implementation of precision medicine. Curr. Probl. Cancer 2017, 41, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.-P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Mullard, A. Use of personalized cancer drugs runs ahead of the science. Nat. News 2015. [Google Scholar] [CrossRef]
- Loehr, M.; Kordes, M.; Malgerud, L.; Kaduthanam, S.; Frödin, J.E.; Karimi, M.; Yachnin, J.; Moro, C.F.; Ghazi, S.; Heuchel, R.L.; et al. Applying evidence-based software for NGS in pancreatic cancer: First results from the PePaCaKa study. JCO 2018, 36, e16214. [Google Scholar] [CrossRef]
- Zimmer, K.; Kocher, F.; Spizzo, G.; Salem, M.; Gastl, G.; Seeber, A. Treatment According to Molecular Profiling in Relapsed/Refractory Cancer Patients: A Review Focusing on Latest Profiling Studies. Comput. Struct. Biotechnol. J. 2019, 17, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Home Page-FP7-Research-Europa. Available online: https://ec.europa.eu/research/fp7/index_en.cfm (accessed on 20 December 2018).
- Homepage. IMI Innovative Medicines Initiative. Available online: http://www.imi.europa.eu/ (accessed on 20 December 2018).
- Cancer Core Europe. Cancer Core Europe. Available online: https://www.cancercoreeurope.eu/ (accessed on 20 December 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NGS Panel Name | Number of Genes | Histology | Provider |
|---|---|---|---|
| EngineusPANEL 600+ | 600+ | Agnostic | Molecular Health GmbH |
| MSK-IMPACT + | 468 | Agnostic | Memorial Sloan Kettering Cancer Center |
| FoundationOne CDx o | 324 | Agnostic C1 | Foundation Medicine, Inc. |
| Oncomine Dx Target Test o | 23 | Specific C2 | ThermoFisher |
| FoundationFocus CDxBRCA Assay o | 2 | Specific C3 | Foundation Medicine, Inc. |
| Praxis Extended RAS Panel o | 2 | Specific C4 | Illumina |
| Name | NCT Number | Enrollment | Start | End | Condition (Disease) | Sponsor |
|---|---|---|---|---|---|---|
| IMPACT II | NCT02152254 | 391 A,* | 2014 A,+ | 2020 E,+ | Metastatic Cancer | MDACC C |
| MPACT | NCT01827384 | 700 E,* | 2013 A,+ | 2019 E,+ | Advanced Malignant Solid Neoplasm | NCI |
| SHIVA | NCT01771458 | 742 A,* | 2012 A,+ | 2016 E,+ | Recurrent/Metastatic Solid Tumor | Institute Curie |
| Name | NCT Number | Enrollment | Start | End | Condition (Disease) | Phase | Sponsor |
|---|---|---|---|---|---|---|---|
| BATTLE2 M1 | NCT01248247 | 334 A,* | 2011 A,+ | 2019 E,+ | NSCLC | 2 | MDACC C1 |
| ALCHEMIST M2 | NCT02194738 | 8300 E,* | 2014 A,+ | 2021 E,+ | NSCLC | 3 | NCI |
| LungMAP M2 | NCT02154490 | 10000 E,* | 2014 A,+ | 2022 E,+ | Squamous Cell Lung Cancer | 2|3 | SOG C2 |
| SAFIR2 Lung M2 | NCT02117167 | 650 E,* | 2014 A,+ | 2022 E,+ | NSCLC | 2 | UNICANCER C3 |
| SAFIR2 Breast M2 | NCT02299999 | 1460 E,* | 2014 A,+ | 2022 E,+ | Breast Cancer | 2 | UNICANCER C4 |
| Name | NCT Number | Enrollment | Start | End | Condition (Disease) |
|---|---|---|---|---|---|
| MATCH M1 | NCT02465060 S1 | 6452 E,* | 2015 A,+ | 2022 E,+ | Advanced Refractory Solid Tumors, Lymphomas, Multiple Myeloma |
| WINTHER M1 | NCT01856296 S2,C1 | 200 E,* | 2013 A,+ | 2018 E,+ | Metastatic Cancer, Advanced Malignancies |
| MOSCATO M2 | NCT01566019 S2 | 1050 E,* | 2011 A,+ | 2019 E,+ | Metastatic Solid Tumors (Any Localization) |
| TAPUR M2 | NCT02693535 S3,C2 | 2980 E,* | 2016 A,+ | 2021 E,+ | Advanced Solid Tumors, Lymphomas (Non-Hodgkin), Multiple Myeloma |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soldatos, T.G.; Kaduthanam, S.; Jackson, D.B. Precision Oncology—The Quest for Evidence. J. Pers. Med. 2019, 9, 43. https://doi.org/10.3390/jpm9030043
Soldatos TG, Kaduthanam S, Jackson DB. Precision Oncology—The Quest for Evidence. Journal of Personalized Medicine. 2019; 9(3):43. https://doi.org/10.3390/jpm9030043
Chicago/Turabian StyleSoldatos, Theodoros G., Sajo Kaduthanam, and David B. Jackson. 2019. "Precision Oncology—The Quest for Evidence" Journal of Personalized Medicine 9, no. 3: 43. https://doi.org/10.3390/jpm9030043
APA StyleSoldatos, T. G., Kaduthanam, S., & Jackson, D. B. (2019). Precision Oncology—The Quest for Evidence. Journal of Personalized Medicine, 9(3), 43. https://doi.org/10.3390/jpm9030043