
The Clinical and Economic Impact of Inaccurate EGFR Mutation Tests in the Treatment of Metastatic Non-Small Cell Lung Cancer

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Patient Population
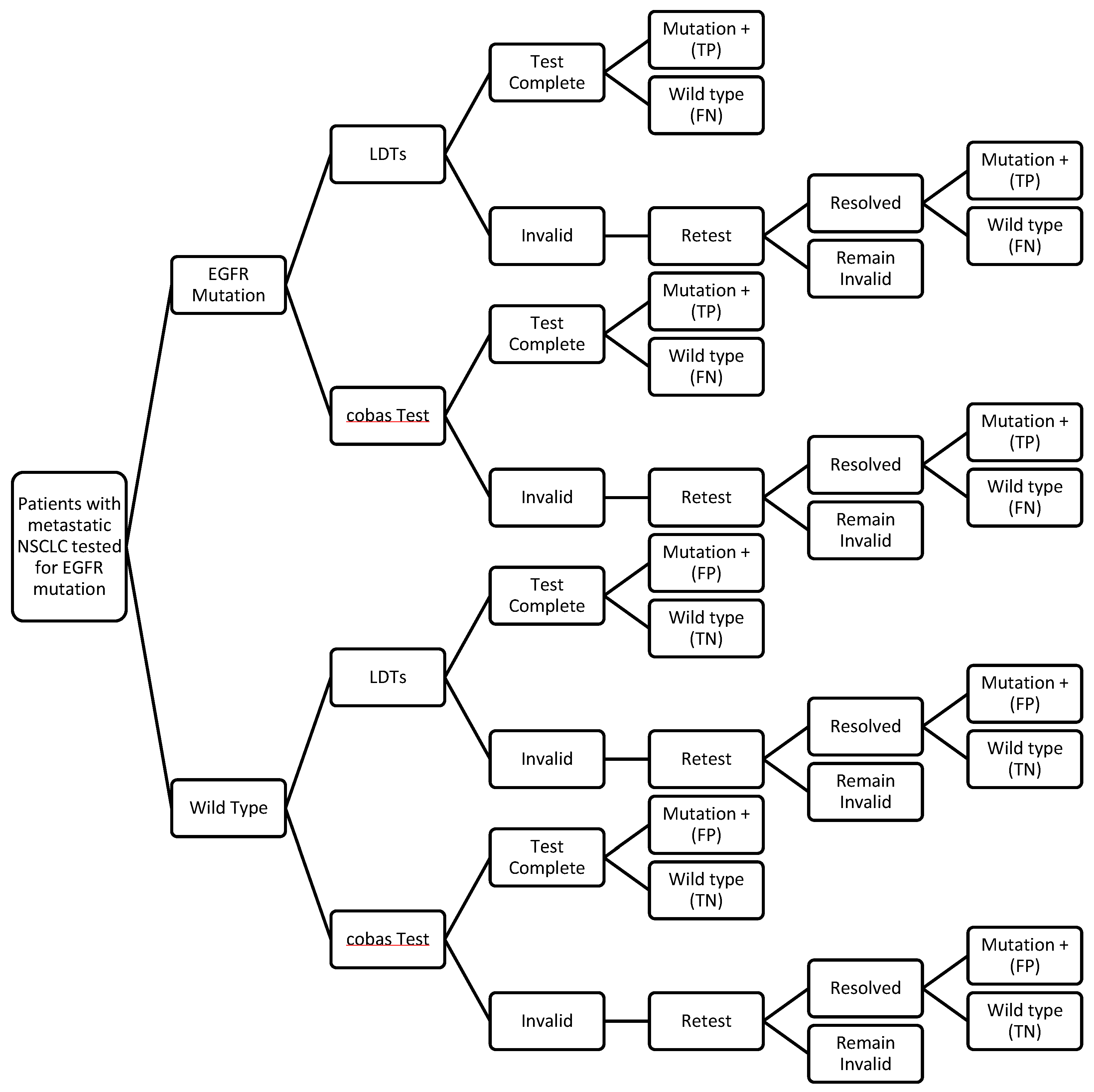
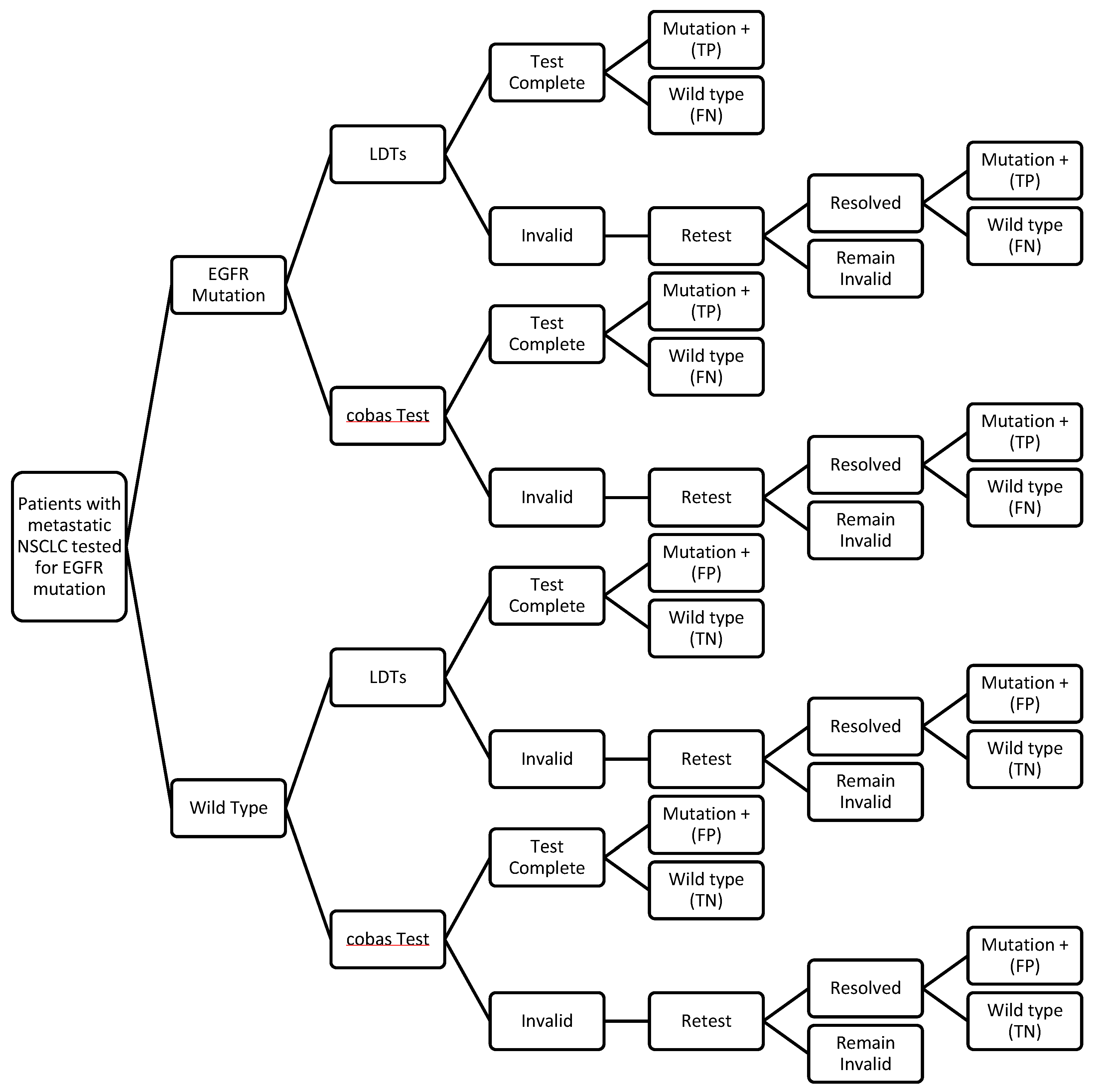
2.3. Decision Analytic Model
2.4. Test Performance
2.5. Clinical Inputs
2.6. Cost Inputs
2.7. Scenario Analyses
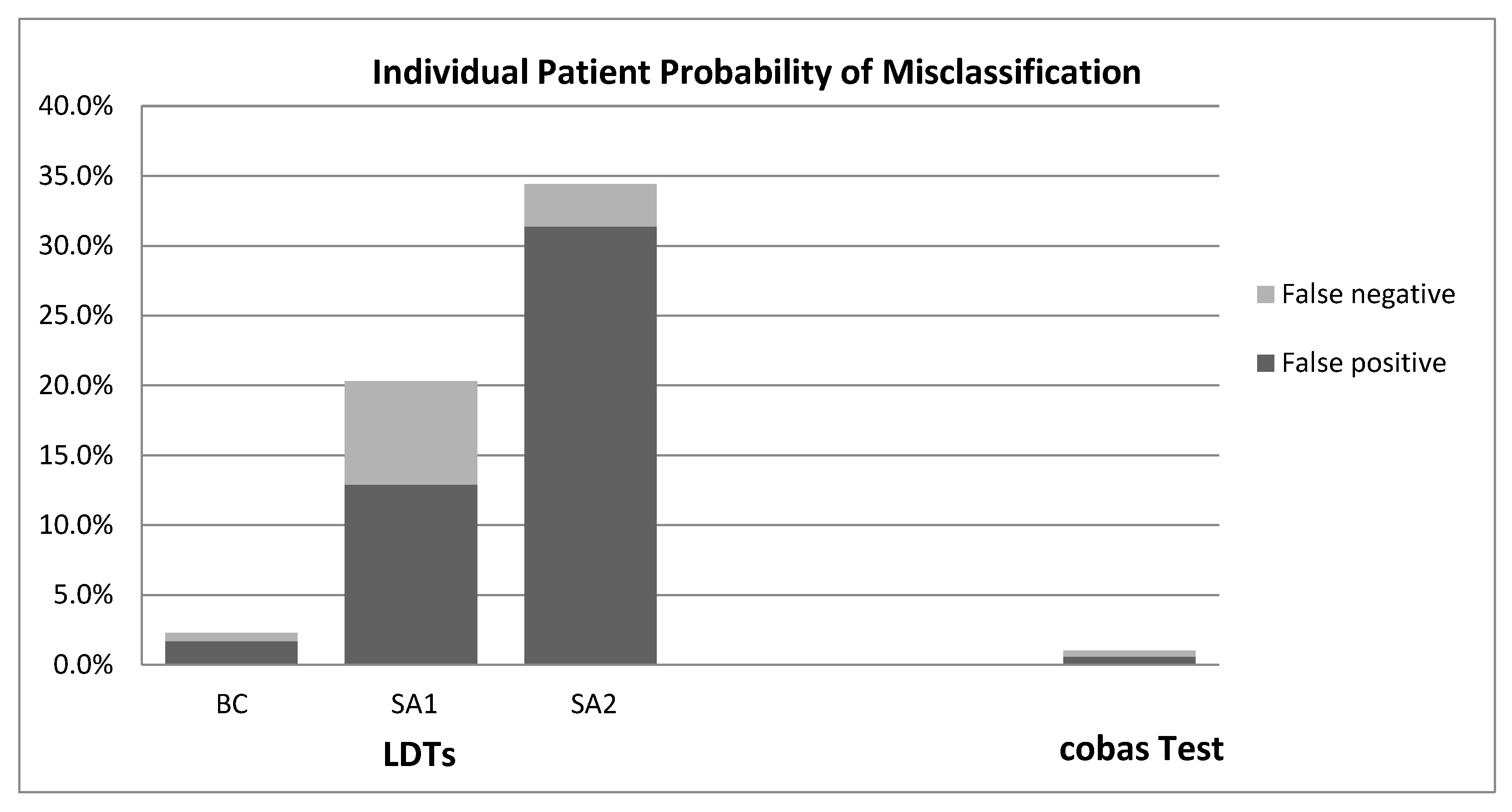
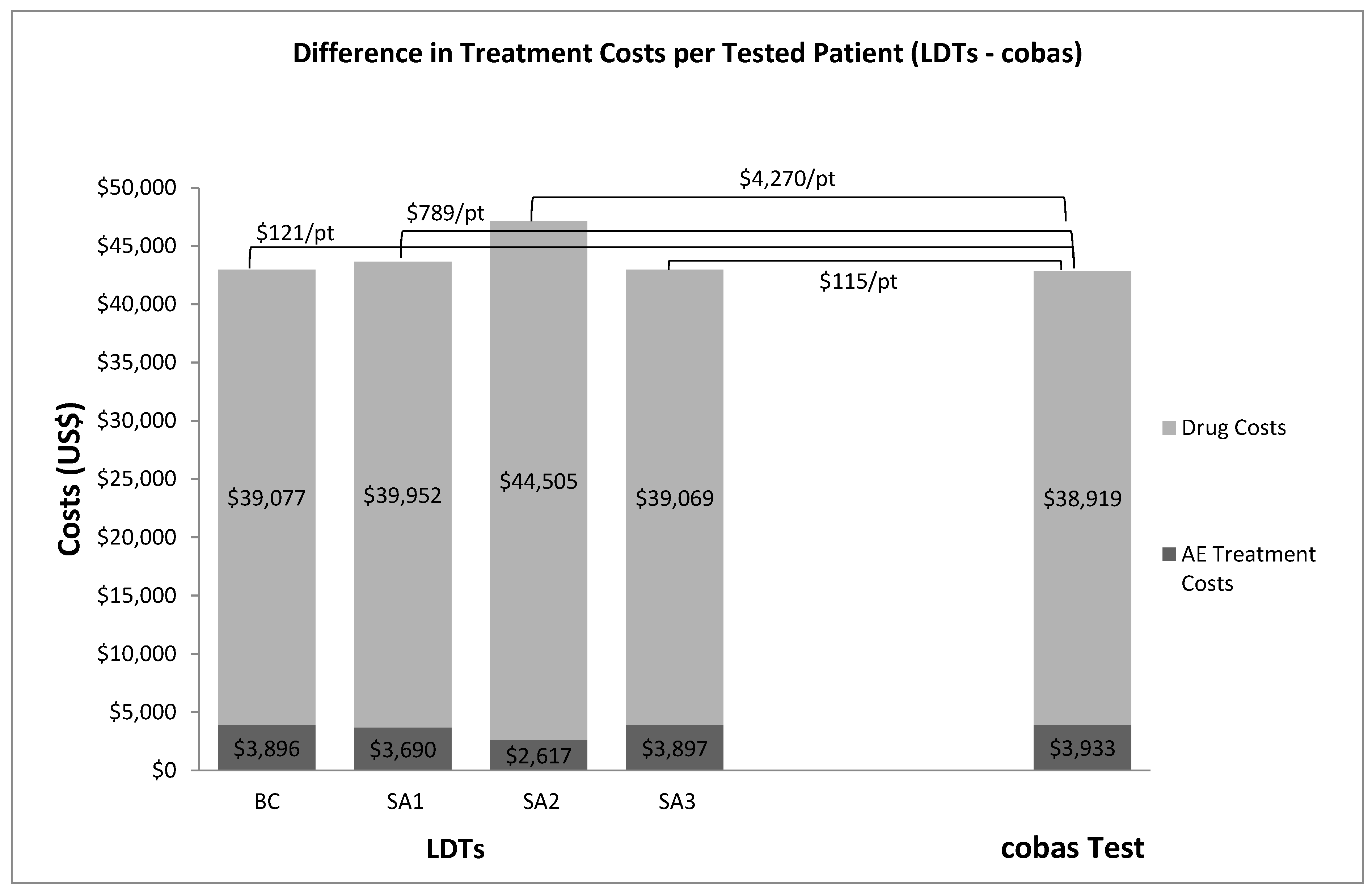
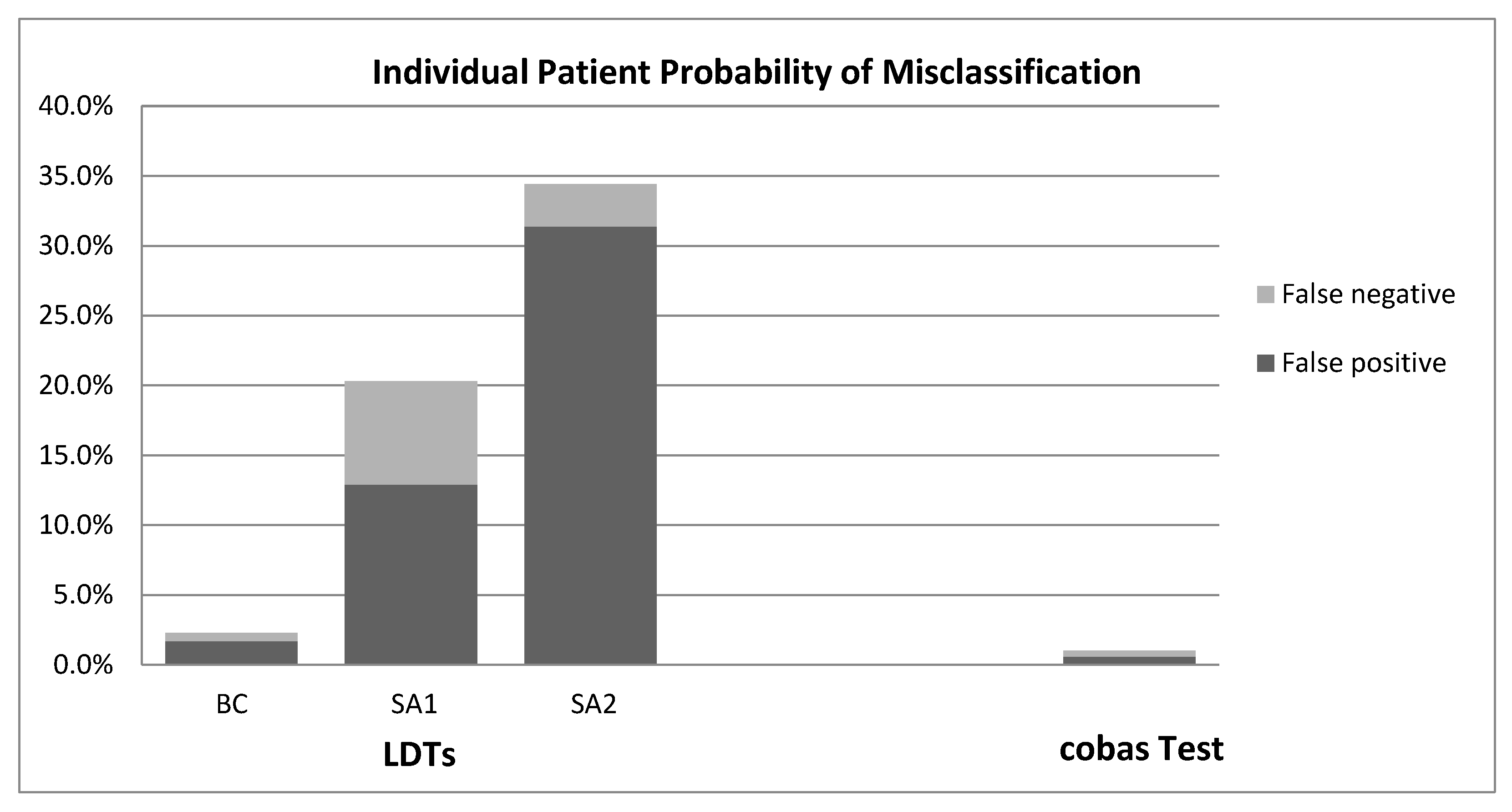
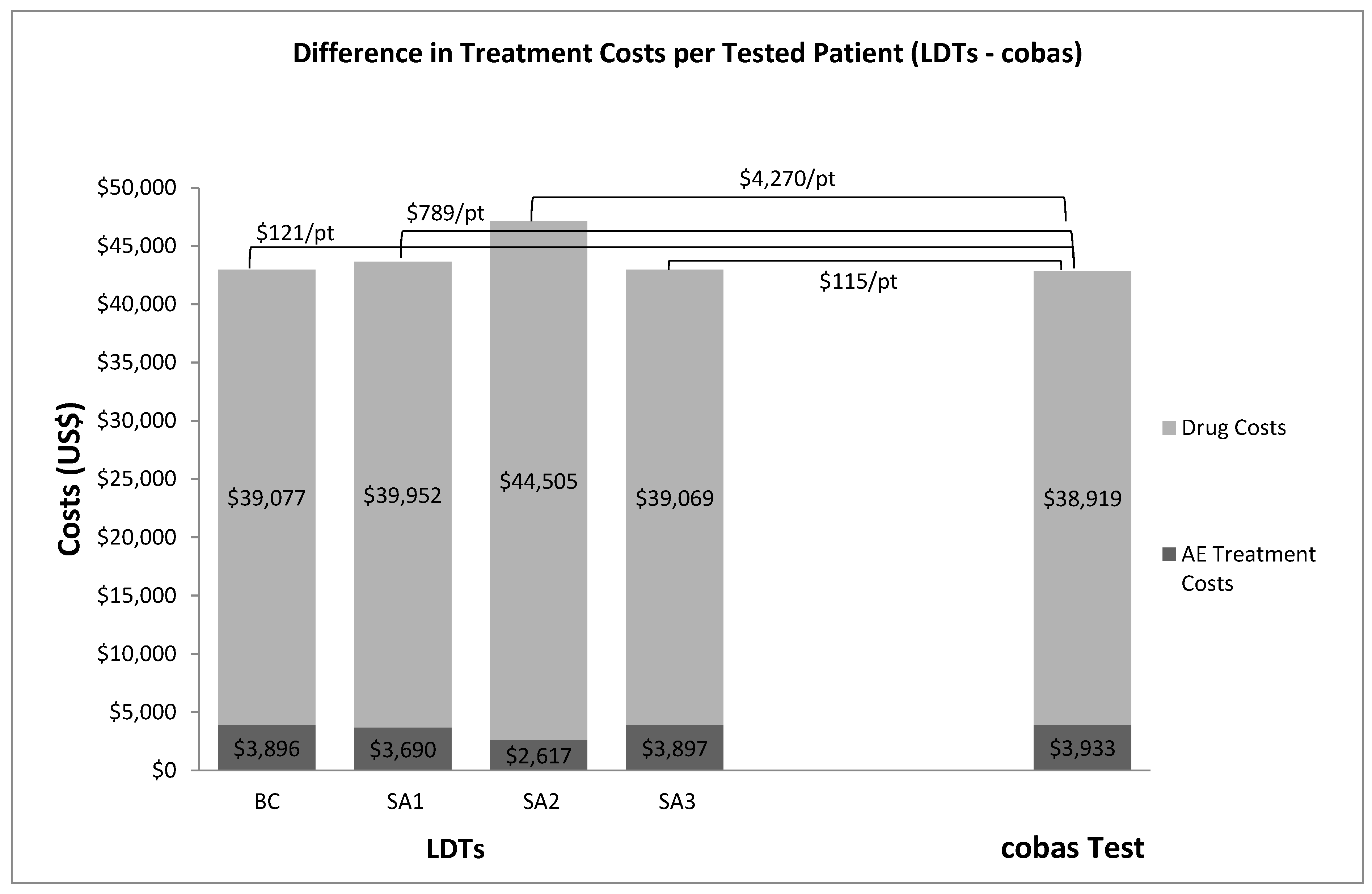
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Balogh, E.P.; Miller, B.T.; Ball, J.R. Improving Diagnosis in Health Care; The National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Hayes, D.F. Considerations for implementation of cancer molecular diagnostics into clinical care. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Garfield, S.; Polisena, J.; Spinner, D.S.; Postulka, A.; Lu, C.Y.; Tiwana, S.K.; Faulkner, E.; Poulios, N.; Zah, V.; Longacre, M. Health technology assessment for molecular diagnostics: Practices, challenges, and recommendations from the Medical Devices and Diagnostic Special Interest Group. Value Health 2016, 19, 577–587. [Google Scholar] [CrossRef] [PubMed]
- The Public Health Evidence for FDA Oversight of Laboratory Developed Tests: 20 Case Studies. US Food and Drug Administration, 2015. Available online: http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UCM472777.pdf (accessed on 12 April 2016).
- Rohr, U.-P.; Binder, C.; Dieterle, T. The value of in vitro diagnostic testing in medical practice: A status report. PLoS ONE 2016, 11, e0149856. [Google Scholar] [CrossRef] [PubMed]
- Laurie, S.A.; Goss, G.D. Role of epidermal growth factor receptor inhibitors in epidermal growth factor receptor wild-type non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer, Version 4. 2016. Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 12 April 2016).
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.-S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors; Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2013, 137, 828–860. [Google Scholar] [PubMed]
- Lynch, J.A.; Khoury, M.J.; Borzecki, A.; Cromwell, J.; Hayman, L.L.; Ponte, P.R.; Miller, G.A.; Lathan, C.S. Epidermal growth factor receptor (EGFR) test utilization in the United States: A case study of T3 translational research. Genet. Med. 2013, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Ardakani, N.M.; Giardina, T.; Grieu-Iacopetta, F.; Tesfai, Y.; Carrello, A.; Taylor, J.; Robinson, C.; Spagnolo, D.; Amanuel, B. Detection of epidermal growth factor receptor mutations in lung adenocarcinoma: Comparing cobas 4800 EGFR assay with Sanger bidirectional sequencing. Clin. Lung Cancer 2016, 17, e113–e119. [Google Scholar] [CrossRef] [PubMed]
- Newman-Toker, D.E.; Pronovost, P.J. Diagnostic errors—The next frontier for patient safety. JAMA 2009, 301, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Commercial Serodiagnostic Tests for Diagnosis of Tuberculosis, Policy Statement. World Health Organization, 2011. Available online: http://apps.who.int/iris/bitstream/10665/44652/1/9789241502054_eng.pdf (accessed on 12 April 2016).
- Benlloch, S.; Botero, M.L.; Beltran-Alamillo, J.; Mayo, C.; Gimenez-Capitán, A.; de Aguirre, I.; Queralt, C.; Ramirez, J.L.; Ramón, y.; Cajal, S.; et al. Clinical validation of a PCR assay for the detection of EGFR mutations in non-small-cell lung cancer: Retrospective testing of specimens from the EURTAC trial. PLoS ONE 2014, 9, e89518. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicenter, open-label, randomized phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures. 2015. Available online: http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf (accessed on 2 May 2016).
- Mavroudis-Chocholis, O.; Ayodele, L. Disease Landscape and Forecast: Non-Small-Cell Lung Cancer; Decision Resources Group Reports, Decision Resources Group: Burlington, MA, USA, 2016. [Google Scholar]
- Boehringer Ingelheim. An International Survey Assessed EGFR Mutation Testing Rates and Treatment Practices in a Specific Type of Lung Cancer. 2015. Available online: http://us.boehringer-ingelheim.com/content/dam/internet/opu/us_EN/documents/Media_Press_Releases/2015/Kantar-Health-Survey-Infographic.pdf (accessed on 14 March 2016).
- Scagliotti, G.V.; Kortsik, C.; Dark, G.G.; Price, A.; Manegold, C.; Rosell, R.; O’brien, M.; Peterson, P.M.; Castellano, D.; Selvaggi, G.; et al. Pemetrexed combined with oxaliplatin or carboplatin as first-line treatment in advanced non-small cell lung cancer: A multicenter, randomized, phase II trial. Clin. Cancer Res. 2005, 11, 690–696. [Google Scholar] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Nafees, B.; Stafford, M.; Gavriel, S.; Bhalla, S.; Watkins, J. Health state utilities for non-small cell lung cancer. Health Qual. Life Outcomes 2008, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.; van Hanswijck de Jonge, P.; Doyle, S.; Cornes, P. Health state utility scores for cancer-related anemia through societal and patient valuations. Value Health 2008, 11, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, C.; Letarte, N.; Mathurin, K.; Yelle, L.; Lachaine, J. A global economic model to assess the cost-effectiveness of new treatments for advanced breast cancer in Canada. J. Med. Econ. 2016, 19, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.J.; Garrison, L.P.; Ramsey, S.D.; Veenstra, D.L. The potential clinical and economic outcomes of pharmacogenomic approaches to EGFR-tyrosine kinase inhibitor therapy in non-small-cell lung cancer. Value Health 2009, 12, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Beausterien, K.M.; Davies, J.; Leach, M.; Meiklejohn, D.; Grinspan, J.L.; O’Toole, A.; Bramham-Jones, S. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: A cross-sectional utility study. Health Qual. Life Outcomes 2010, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- U.S. Centers for Disease Control and Prevention [Internet]. National Center for Health Statistics, Body Measurements. Available online: http://www.cdc.gov/nchs/fastats/body-measurements.htm (accessed on 15 March 2016).
- Sacco, J.J.; Botten, J.; Macbeth, F.; Bagust, A.; Clark, P. The average body surface area of adult cancer patients in the UK: A multicenter retrospective study. PLoS ONE 2010, 5, e8933. [Google Scholar] [CrossRef] [PubMed]
- Centers for Medicare & Medicaid Services. 2015 ASP Drug Pricing Files (July). Available online: https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice/2015ASPFiles.htmlCMS (accessed on 12 May 2016).
- Revenue Cycle Inc. 2016 Billing and Coding Update for Radiation and Medical Oncology. 29 January 2016. Available online: http://www.cancerexecutives.org/assets/docs/members-only/2016%20billing%20coding%20update%20for%20rad%20med%20onc%201%2029%20161.pdf (accessed on 12 May 2016).
- Agency for Healthcare Research and Quality. HCUPnet. Available online: http://hcupnet.ahrq.gov/ (accessed on 10 May 2016).
- AABB. 2016 Medicare Proposed Hospital Outpatient Payments. Available online: https://www.aabb.org/advocacy/reimbursementinitiatives/Documents/2016-HOPPS-Proposed-Rule-Summary.pdf (accessed on 31 March 2016).
- Sharfstein, J. FDA regulation of laboratory-developed diagnostic tests, protect the public, advance the science. JAMA 2015, 313, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Westwood, M.E.; Joore, M.A.; Whiting, P.; van Asselt, T.; Armstrong, N.; Lee, K.; Misso, K.; Kleijnen, J.; Severens, H. Epidermal growth factor receptor tyrosine kinase (EGFR-TK) mutation testing in adults with locally advanced or metastatic non-small-cell lung cancer: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2014, 18, 1–166. [Google Scholar] [CrossRef] [PubMed]
- Patton, S.; Normanno, N.; Blackhall, F.; Murray, S.; Kerr, K.M.; Dietel, M.; Filipits, M.; Benlloch, S.; Popat, S.; Stahel, R. Assessing standardization of molecular testing for non-small-cell lung cancer: Results of a worldwide external quality assessment (EQA) scheme for EGFR mutation testing. Br. J. Cancer 2014, 111, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Deans, Z.C.; Bilbe, N.; O’Sullivan, B.; Lazarou, L.P.; de Castro, D.G.; Parry, S.; Dodson, A.; Taniere, P.; Clark, C.; Butler, R. Improvement in the quality of molecular analysis of EGFR in non-small-cell lung cancer detected by three rounds of external quality assessment. J. Clin. Pathol. 2013, 66, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Vyberg, M.; Nielsen, S.; Røge, R.; Sheppard, B.; Ranger-Moore, J.; Walk, E.; Gartemann, J.; Rohr, U.-P.; Teichgräber, V. Immunohistochemical expression of HER2 in breast cancer: Socioeconomic impact of inaccurate tests. BMC Health Serv. Res. 2015, 15, 352. [Google Scholar] [CrossRef] [PubMed]
- Garrison, L.P.; Babigumira, J.B.; Masaquel, A.; Wang, B.C.M.; Lalla, D.; Brammer, M. The lifetime economic burden of inaccurate HER2 testing: Estimating the costs of false-positive and false-negative HER2 test results in US patients with early-stage breast cancer. Value Health 2015, 18, 541–546. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Estimate | Reference |
|---|---|---|
| New lung cancer cases diagnosed in US in 2015 | 221,200 | [15] |
| % Lung cancer cases that are NSCLC | 85% | [16] |
| % NSCLC that are adenocarcinoma, large cell, or unspecified histology | 73% | [16] |
| % NSCLC advanced or metastatic at diagnosis (Stage IIIb or IV) | 58% | [16] |
| Diagnosed Cohort | 79,608 | |
| % Diagnosed patients tested for EGFR mutation in US | 76% | [17] |
| National Analytic Cohort | 60,502 | |
| Prevalence of EGFR mutation in US | 19% | [16] |
| Cobas Test Results | Mutation Status Based on MPP Resolution | ||
| Mutation + | Wild Type | Total | |
| Mutation + | 151 | 2 | 153 |
| Wild Type | 3 | 276 | 279 |
| Total | 154 | 278 | 432 |
| Sensitivity: 98.1% Specificity: 99.3% Invalid rate: 8.9% | |||
| LDT Results | Mutation Status based on MPP Resolution | ||
| Mutation + | Wild Type | Total | |
| Mutation + | 149 | 6 | 155 |
| Wild Type | 5 | 272 | 277 |
| Total | 154 | 278 | 432 |
| Sensitivity: 96.8% Specificity: 97.8% Invalid rate: 15.6% | |||
| Survival | Chemotherapy (Carboplatin + Pemetrexed) [18] | Erlotinib (EGFR Mutation Positive) [14] | Erlotinib (EGFR Wild Type) [14] |
|---|---|---|---|
| Mean duration of PFS (months) | 8.55 | 11.50 | 2.98 [19] |
| Grade 3–4 Adverse Events | |||
| Anemia | 7.7% | ||
| Arthralgia | 1.0% | 1.0% | |
| Diarrhea | 5.0% | 5.0% | |
| Fatigue | 7.7% | 6.0% | 6.0% |
| Febrile Neutropenia | 5.1% | ||
| Infection | 2.6% | ||
| Neuropathy | 1.0% | 1.0% | |
| Neutropenia | 25.6% | ||
| Pneumonitis | 1.0% | 1.0% | |
| Rash | 13.0% | 13.0% | |
| Stomatitis | 2.6% | ||
| Thrombocytopenia | 17.9% |
| Adverse Event | Utility Estimate | Reference |
|---|---|---|
| Base-line, stable disease, no toxicity | 0.653 | [20] |
| Anemia (grade 3–4) | 0.583 | [21] |
| Arthralgia (grade 3–4 ) | 0.589 | [22] |
| Diarrhea (grade 3–4 ) | 0.606 | [20] |
| Fatigue (grade 3–4 ) | 0.580 | [20] |
| Febrile Neutropenia (grade 3–4 ) | 0.563 | [20] |
| Infection (grade 3–4 ) | 0.423 | [22] |
| Neuropathy (grade 3–4 ) | 0.620 | [23] |
| Neutropenia (grade 3–4 ) | 0.563 | [20] |
| Pneumonitis (grade 3–4 ) | 0.560 | [24] |
| Rash (grade 3–4 ) | 0.621 | [20] |
| Stomatitis (grade 3–4 ) | 0.610 | [23] |
| Thrombocytopenia (grade 3–4 ) | 0.545 | [22] |
| Drug | Administration Route | Unit Cost | Dose per Cycle | Drug Cost per Cycle | Drug Admin. Cost per Cycle | Duration of Treatment | Total Cost |
|---|---|---|---|---|---|---|---|
| Carboplatin | IV infusion | $3.85/50 mg | 669.7 mg | $51.57 | $136.55 | 6 cycles (median) | $1,129 |
| Pemetrexed | IV infusion | $61.33/10 mg | 895 mg | $5,489.04 | $63.24 | 6 cycles (median) | $33,314 |
| Folic acid * | Oral | Supplements not reimbursed by Medicare | Assume negligible | ||||
| Vitamin B12 * | Subcutaneous injection | $4.39/1 mg | $53.52 | 3 total injections | $174 | ||
| Dexamethasone * | Oral | $0.25/0.25 mg | 24 mg | $24.00 | 6 cycles (median) | $144 | |
| Ondansetron * | Subcutaneous injection | $0.073/1 mg | 24 mg | $1.75 | $53.52 | 6 cycles (median) | $332 |
| Total Chemotherapy and Admin. Cost per Patient | $35,092 | ||||||
| Total Erlotinib Cost per Patient | Oral | $223.63/150 mg | 150 mg/day | 246 days (median) | $55,013 |
| Adverse Event | Setting of Care/Primary Treatment Procedure | Treatment Cost Estimate (2015$) | Reference |
|---|---|---|---|
| Anemia | Outpatient/Blood transfusion | $598 | [30] |
| Arthralgia * | Clinical opinion | ||
| Diarrhea * | Clinical opinion | ||
| Fatigue * | Clinical opinion | ||
| Febrile Neutropenia | Inpatient | $20,254 | [29] |
| Infection | Inpatient | $16,657 | [29] |
| Neuropathy * | Clinical opinion | ||
| Neutropenia | Outpatient/Neupogen | $12,423 | [27,30] |
| Pneumonitis | Inpatient | $14,097 | [29] |
| Rash * | Clinical opinion | ||
| Thrombocytopenia | Outpatient/Blood transfusion | $795 | [30] |
| Sensitivity | Specificity | Invalid Rate | Reference | |
|---|---|---|---|---|
| cobas test | 98.1% | 99.3% | 8.9% | [13] |
| LDTs-Base-Case (“BC”) | 96.8% | 97.8% | 15.6% | [13] |
| LDTs-Scenario 1 (“SA1”) | 61.0% | 84.0% | 15.6% | [32] |
| LDTs-Scenario 2 (“SA2”) | 84.0% | 61.0% | 15.6% | [32] |
| LDTs-Scenario 3 (“SA3”) | 96.8% | 97.8% | 20.0% | Exploratory |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.M.; Palma, J.F.; Scudder, S.; Poulios, N.; Liesenfeld, O. The Clinical and Economic Impact of Inaccurate EGFR Mutation Tests in the Treatment of Metastatic Non-Small Cell Lung Cancer. J. Pers. Med. 2017, 7, 5. https://doi.org/10.3390/jpm7030005
Cheng MM, Palma JF, Scudder S, Poulios N, Liesenfeld O. The Clinical and Economic Impact of Inaccurate EGFR Mutation Tests in the Treatment of Metastatic Non-Small Cell Lung Cancer. Journal of Personalized Medicine. 2017; 7(3):5. https://doi.org/10.3390/jpm7030005
Chicago/Turabian StyleCheng, Mindy M., John F. Palma, Sidney Scudder, Nick Poulios, and Oliver Liesenfeld. 2017. "The Clinical and Economic Impact of Inaccurate EGFR Mutation Tests in the Treatment of Metastatic Non-Small Cell Lung Cancer" Journal of Personalized Medicine 7, no. 3: 5. https://doi.org/10.3390/jpm7030005
APA StyleCheng, M. M., Palma, J. F., Scudder, S., Poulios, N., & Liesenfeld, O. (2017). The Clinical and Economic Impact of Inaccurate EGFR Mutation Tests in the Treatment of Metastatic Non-Small Cell Lung Cancer. Journal of Personalized Medicine, 7(3), 5. https://doi.org/10.3390/jpm7030005