
Information Technology Support for Clinical Genetic Testing within an Academic Medical Center

 , and
, and 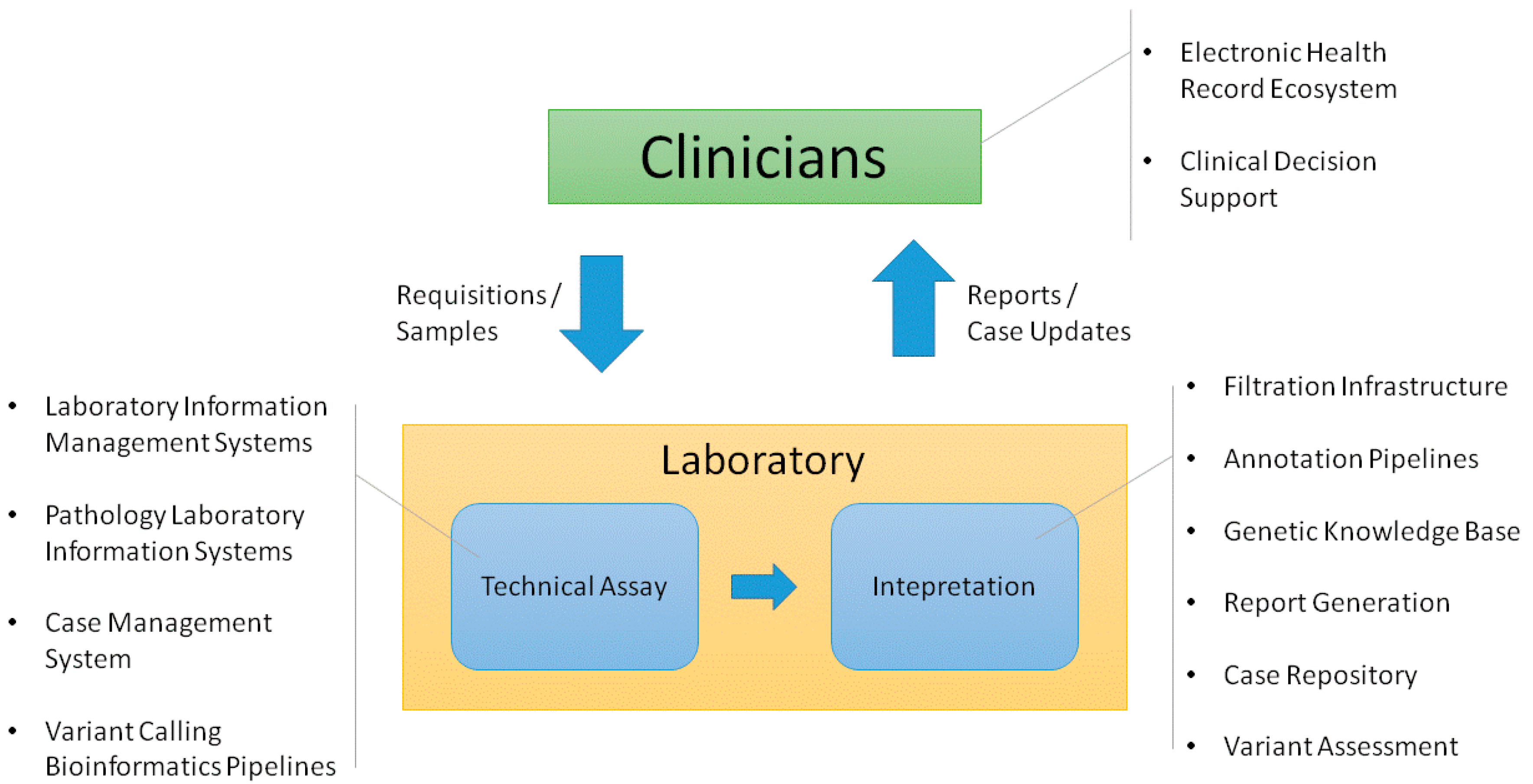{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
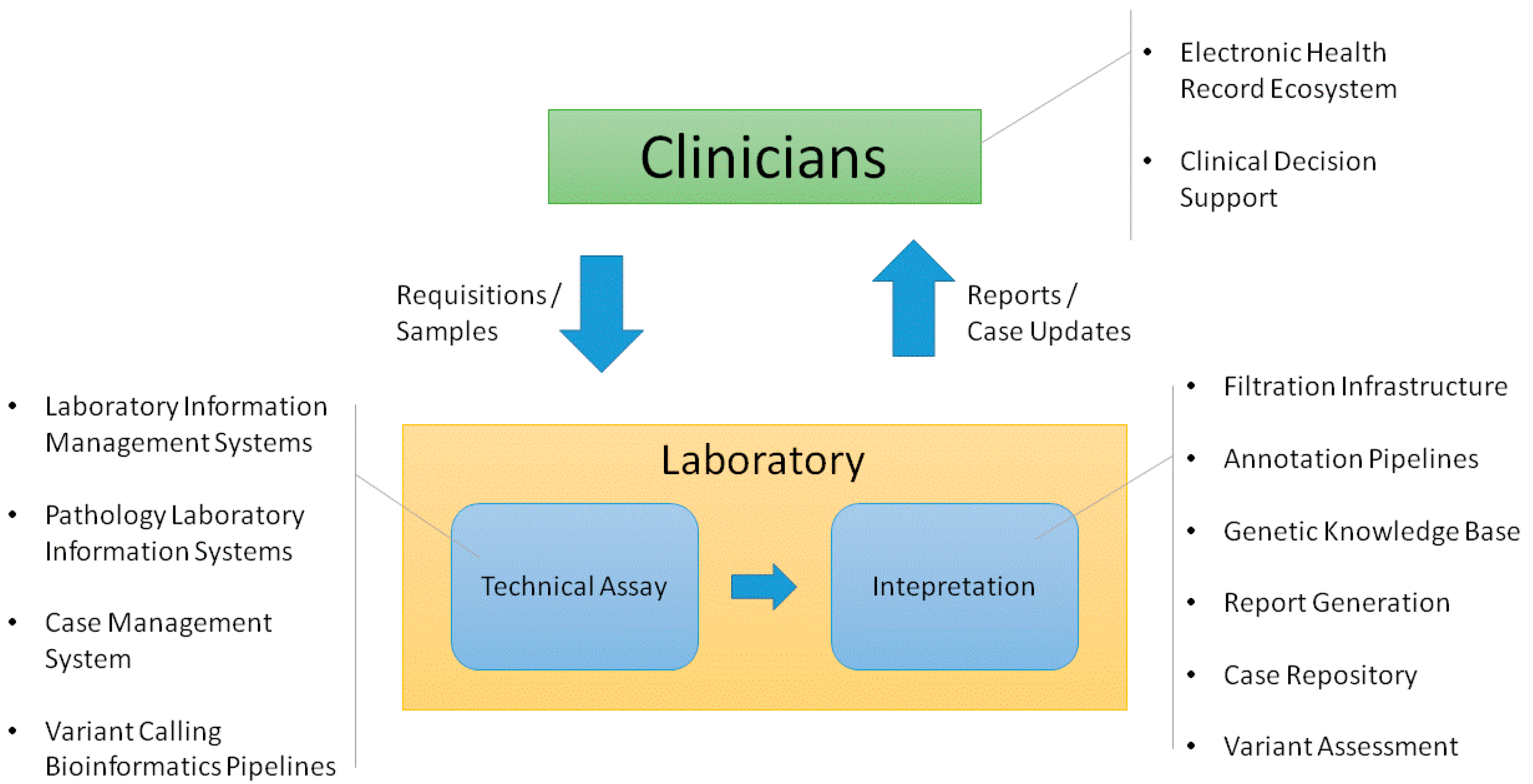
2. Clinician Facing Infrastructure
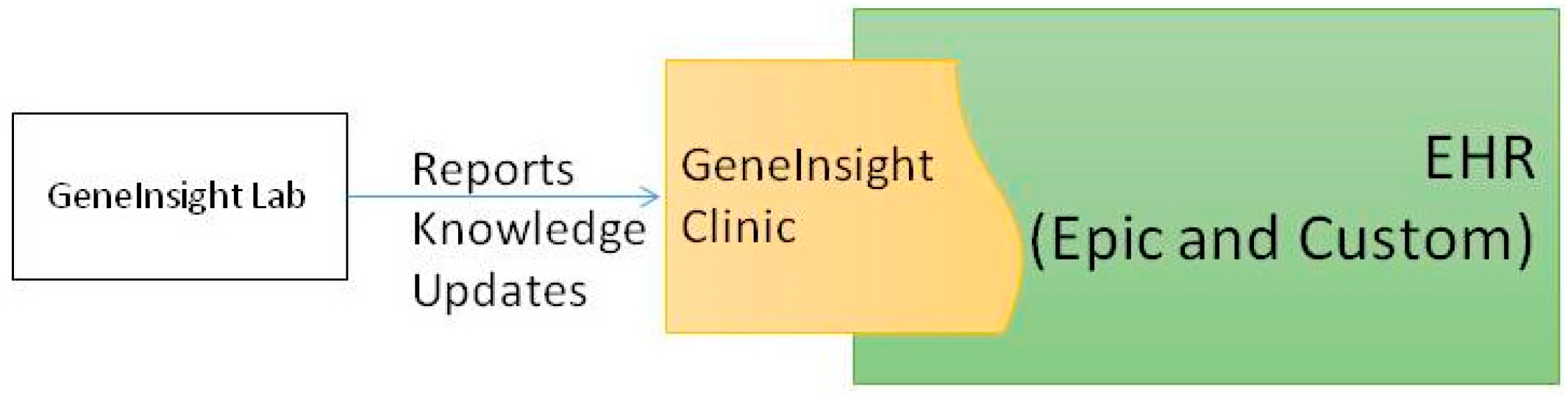
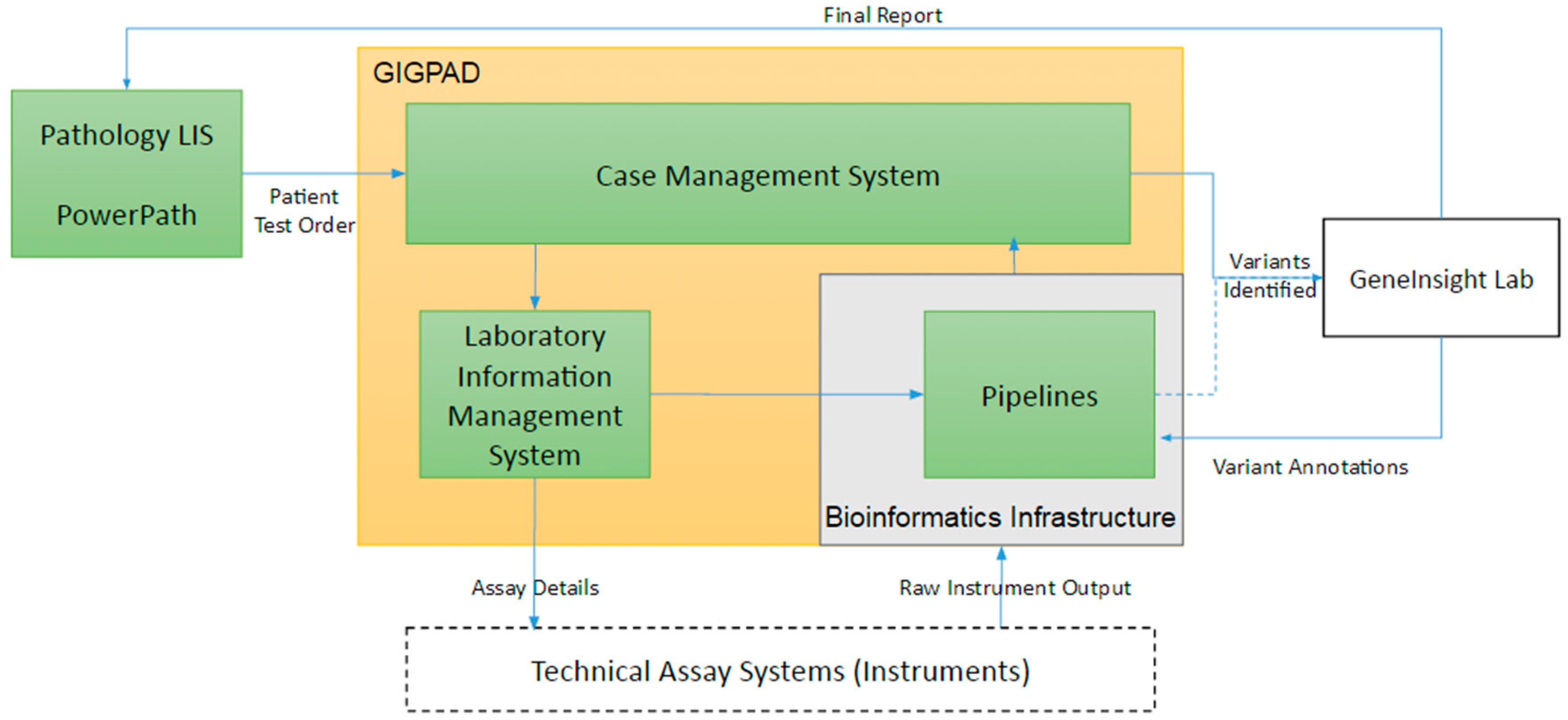
3. Infrastructure Supporting the Technical Assay
3.1. The Technical Assay Workflow
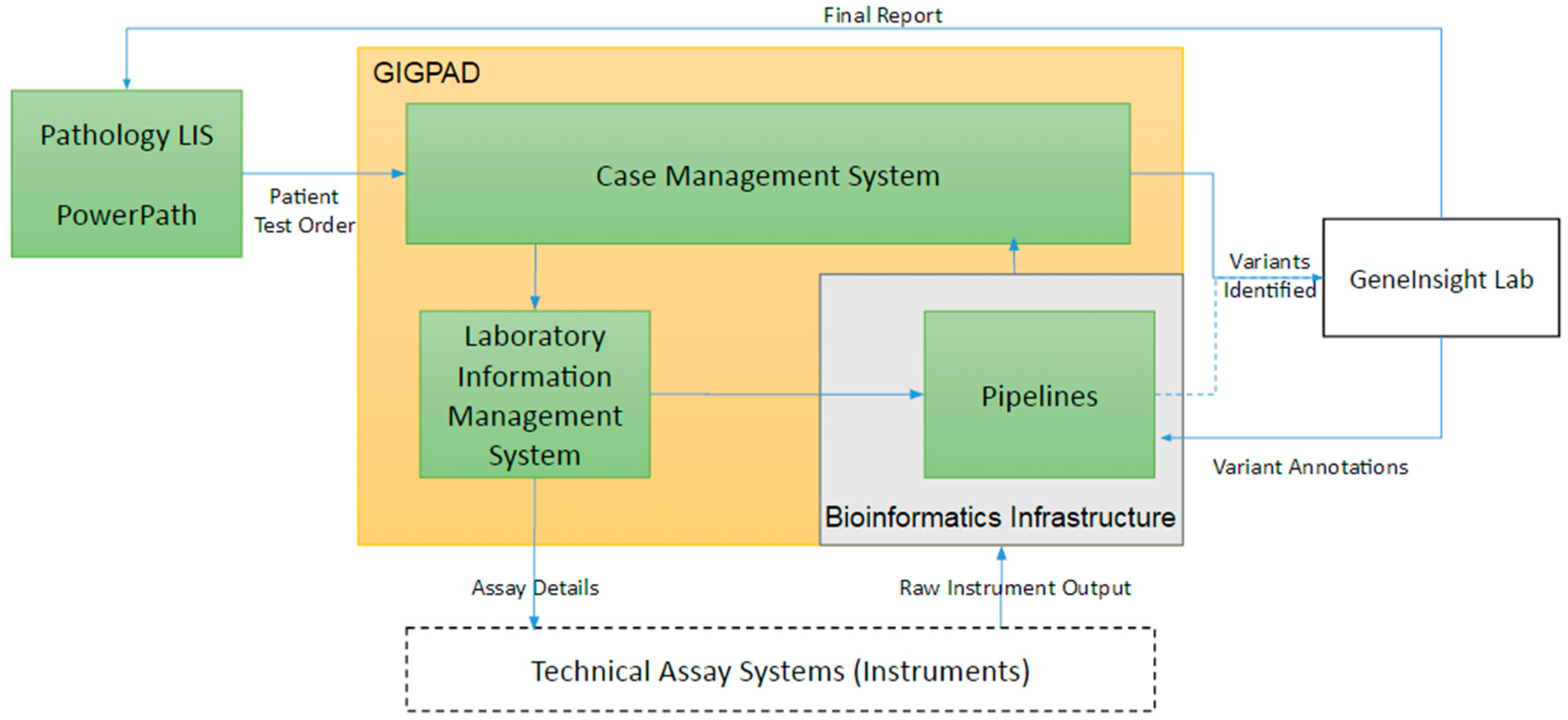
3.2. The Use of Pathology Laboratory Information Systems
3.3. Case Management System (CMS)
3.4. Laboratory Information Management Systems (LIMS)
3.5. Oligo Management
3.6. Variant Calling Bioinformatic Pipelines
4. Infrastructure Supporting Interpretation
4.1. The Interpretation Workflow
4.2. Genetic Knowledge Base and Case Repository
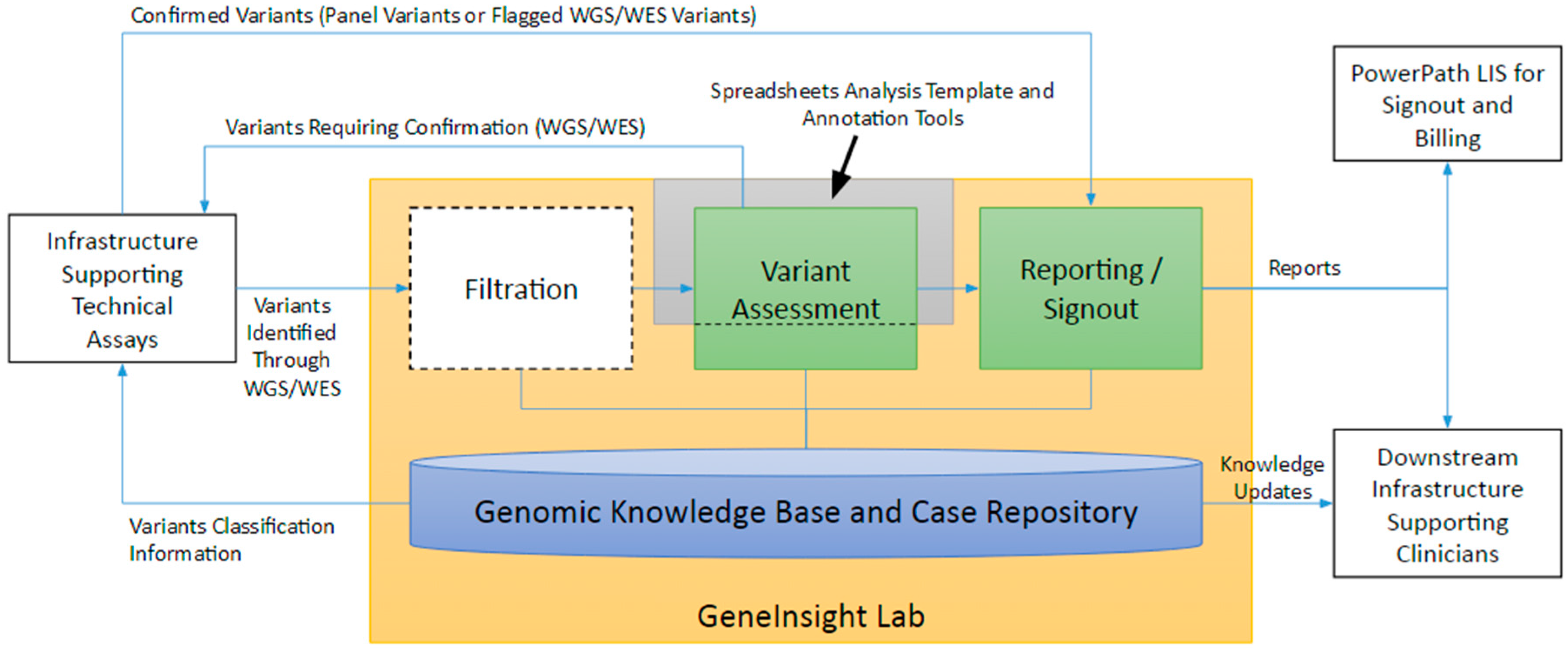
4.3. Filtration
4.4. Variant Assessment
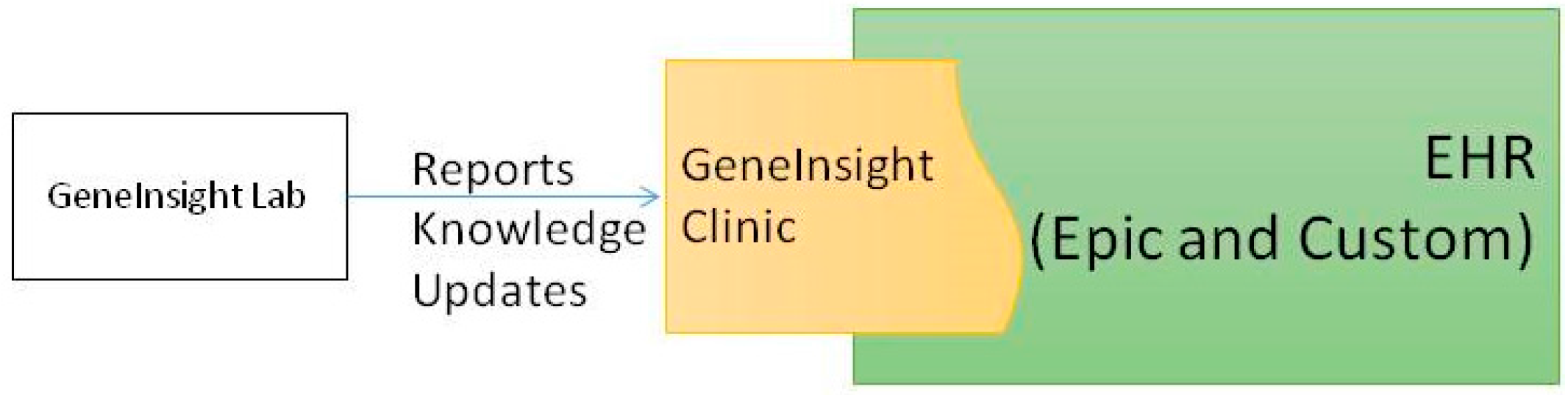
4.5. Reporting and Sign out
5. Cross Institutional Sharing
6. The Future
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aronson, S.J.; Rehm, H.L. Building the foundation for genomics in precision medicine. Nature 2015, 526, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Starren, J.; Williams, M.S.; Bottinger, E.P. Crossing the omic chasm: A time for omic ancillary systems. JAMA 2013, 309, 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Aronson, S.J.; Clark, E.H.; Babb, L.J.; Baxter, S.; Farwell, L.M.; Funke, B.H.; Hernandez, A.L.; Joshi, V.A.; Lyon, E.; Parthum, A.R.; et al. The GeneInsight Suite: A platform to support laboratory and provider use of DNA-based genetic testing. Hum. Mutat. 2011, 32, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Aronson, S.J.; Clark, E.H.; Varugheese, M.; Baxter, S.; Babb, L.J.; Rehm, H.L. Communicating new knowledge on previously reported genetic variants. Genet. Med. 2012, 14, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.A.; Williams, M.S. Electronic medical records and personalized medicine. Hum. Genet. 2011, 130, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Alterovitz, G.; Warner, J.; Zhang, P.; Chen, Y.; Ullman-Cullere, M.; Kreda, D.; Kohane, I.S. SMART on FHIR Genomics: Facilitating standardized clinico-genomic apps. J. Am. Med. Inform. Assoc. 2015, 22, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 11.10.1–11.10.33. [Google Scholar]
- Pugh, T.J.; Amr, S.S.; Bowser, M.J.; Gowrisankar, S.; Hynes, E.; Mahanta, L.M.; Rehm, H.L.; Funke, B.; Lebo, M.S. VisCap: Inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Duzkale, H.; Shen, J.; McLaughlin, H.; Alfares, A.; Kelly, M.A.; Pugh, T.J.; Funke, B.H.; Rehm, H.L.; Lebo, M.S. A systematic approach to assessing the clinical significance of genetic variants. Clin. Genet. 2013, 84, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—the clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aronson, S.; Mahanta, L.; Ros, L.L.; Clark, E.; Babb, L.; Oates, M.; Rehm, H.; Lebo, M. Information Technology Support for Clinical Genetic Testing within an Academic Medical Center. J. Pers. Med. 2016, 6, 4. https://doi.org/10.3390/jpm6010004
Aronson S, Mahanta L, Ros LL, Clark E, Babb L, Oates M, Rehm H, Lebo M. Information Technology Support for Clinical Genetic Testing within an Academic Medical Center. Journal of Personalized Medicine. 2016; 6(1):4. https://doi.org/10.3390/jpm6010004
Chicago/Turabian StyleAronson, Samuel, Lisa Mahanta, Lei Lei Ros, Eugene Clark, Lawrence Babb, Michael Oates, Heidi Rehm, and Matthew Lebo. 2016. "Information Technology Support for Clinical Genetic Testing within an Academic Medical Center" Journal of Personalized Medicine 6, no. 1: 4. https://doi.org/10.3390/jpm6010004
APA StyleAronson, S., Mahanta, L., Ros, L. L., Clark, E., Babb, L., Oates, M., Rehm, H., & Lebo, M. (2016). Information Technology Support for Clinical Genetic Testing within an Academic Medical Center. Journal of Personalized Medicine, 6(1), 4. https://doi.org/10.3390/jpm6010004