Trends in Personalized Therapies in Oncology: The (Venture) Capitalist’s Perspective

Abstract
:1. Introduction

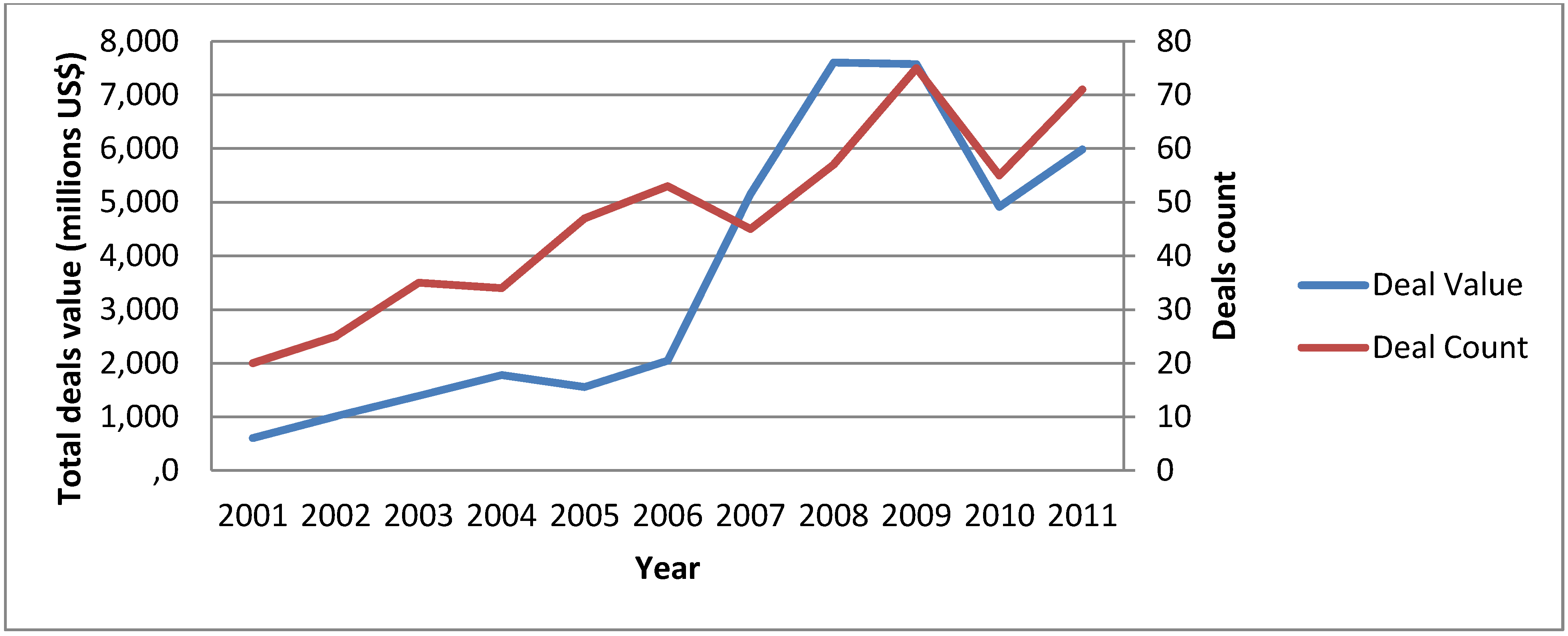{kind=link}
{kind=link}
| Molecule | Target | Biomarker | Indication |
|---|---|---|---|
| vemurafenib | BRAF kinase | BRAF V600E mutant | melanoma |
| vandetanib | VEGFR, EGFR, RETR | not included on the label | thyroid cancer |
| abiraterone | CYP 17A1 | not included on the label | prostate cancer |
| ipilimumab | CTLA-4 | not included on the label | melanoma |
| brentuximab vedotin | CD30 (ACD technology) | not included on the label | lymphoma |
| crizotinib | ALK | ALK-EML4 fusion protein | lung cancer |
| Molecule | Target | Biomarker | Indication |
|---|---|---|---|
| busulfan ab | DNA | Ph+ | CML |
| irinotecan ab | Topoisomerase I | *UGT1A1 | colorectal cancer |
| cetuximab ab | EGFR | EGFR+, KRAS-wt c | colorectal cancer; head and neck cancer |
| imatinib ab | bcr-abl (c-kit, PDGFR) | Ph+, C-Kit+ | CML, GIST |
| transtuzumab ab | ErbB2 | ErbB2 overexpression c | breast cancer; gastrointestinal cancer |
| getifinib b | EGFR | EGFR-TK mutant c | NSCLC |
| tamoxifen ab | ER | ER+ | breast cancer |
| denileukin diftitox a | IL2R | CD25+ c | cutaneous T-cell lymphoma |
| mercaptopurine ab | DNA synthesis | *TPMT | leukemia; non-Hodgkin’s lymphoma |
| dasatinib b | bcr-abl, src | Ph+ | ALL, CML |
| thioguanine ab | DNA | *TPMT | acute leukemia, CLL |
| erlotinib b | EGFR | EGFR+ | NSCLC; pancreatic cancer |
| nilotinib b | bcr-abl (and others) | Ph+ | CML |
| arsenic trioxide b | apoptotic | PML/RAR alpha gene+ | AML |
| lapatinib ab | EGFR, ErbB2 | HER-2+ c | breast cancer |
| panitumumab ab | EGFR | EGFR+, KRAS-wt c | colorectal cancer |
| capecitabine ab | DNA synthesis | *DPD | breast cancer, colorectal cancer |
| aromatase inhibitors abd | estrogen synthesis | *Aromatase+, ER+ | breast cancer |
| DNA intercalators abe | DNA | *Topoisomerase IIα copy number | breast cancer |
2. Global Trends in Oncology Drug Development

2.1. Progressive Shift towards Molecularly Targeted Therapies
2.2. Combining Targeted Therapies
| Company 1 | Company 2 | Compound 1 | Compound 2 | Modality | Indication |
|---|---|---|---|---|---|
| Merck & Co. | AstraZeneca | MK-2206 AKT inhibitor | selumetinib MEK-1 inhibitor | combination | lung cancer |
| Merck Serono | Sanofi | pimasertib MEK-1 inhibitor | SAR245408 PI3K inhibitor | combination | solid tumors |
| BMS | Roche | ipilimumab CTLA4 inhibitor | vemurafenib BRAF inhibitor | combination | melanoma |
2.3. Market Fragmentation
2.4. Use of Biomarkers as Companion Diagnostics
| Amgen | DxS | panitumumab | K-Ras | colorectal cancer |
| Eli Lilly | Genomic Health | cetuximab | mRNA signature | colorectal cancer |
| Ipsen | BioMérieux | several n.d. | several n.d. | breast cancer |
| Merk KGaA | OncoMethylome | cilengitide | MGMT gene methylation | brain cancer |
| Pfizer | Genomic Health | several n.d. | mRNA signature | renal cancer |
| OSI | Dako | ertolinitib hydrochloride | EGFR | NSCLC |
| Merck & Co | OncoMethylome | temozolomide | MGMT gene methylation | brain cancer |
| AstraZeneca | Dako | several n.d. | several n.d. | several |
2.5. Move towards Proof of Relevance versus Proof of Concept
3. How Venture Capital Is Deployed in Personalized Medicine
3.1. The Asset-Centric versus Company-Building Approach
3.2. The ‘New Mode of Action’ versus ‘Fast Follower’
3.3. Why Personalized Medicine Is “Hot” for Venture Capitalists
4. What the Venture Capital/Biopharmaceutical Industry Is Interested in
4.1. A Selective Look at MoA’s Currently in Early/Late Stage Trials
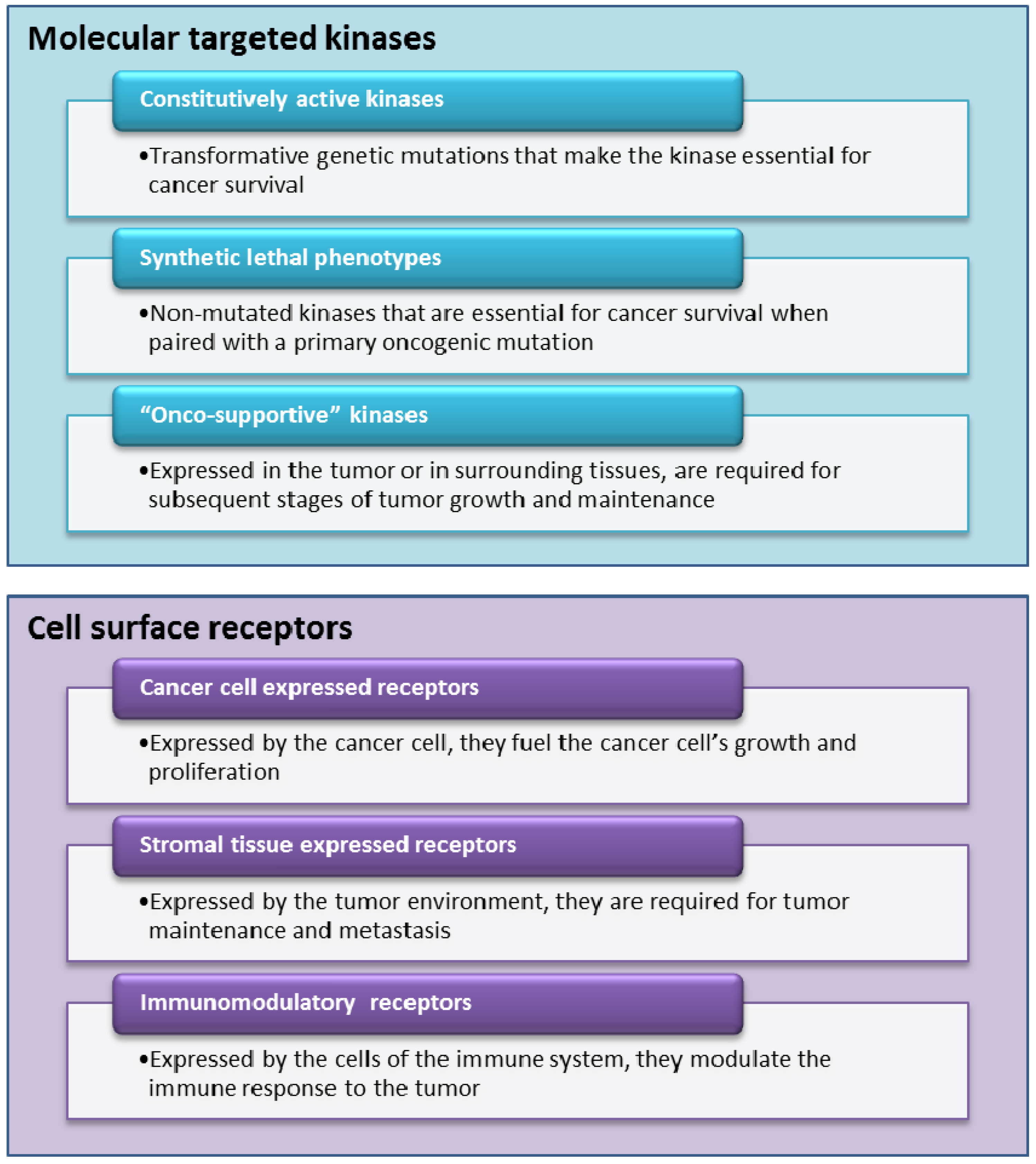
4.1.1. Molecularly Targeted Kinases Inhibitors
4.1.2. Ligands for Cell Surface Receptors
4.2. From Bench to Bedside and Back to Bench
4.3. Molecular Profiling with Whole Genome Sequencing
4.4. Novel Models of Collaboration and Partnerships
5. Conclusions
References
- FDA. Drugs@FDA. Available online: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ (accessed on 7 February 2012).
- Datamonitor. Market and Product Forecasts: Targeted Cancer Therapies—Trend towards Personalized Medicine Will Lead to Market Fragmentation. 2011. HC00118–008.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Hinds, D.A.; Stuve, L.L.; Nilsen, G.B.; Halperin, E.; Eskin, E.; Ballinger, D.G.; Frazer, K.A.; Cox, D.R. Whole-genome patterns of common DNA variation in three human populations. Science 2005, 307, 1072–1079. [Google Scholar] [CrossRef]
- Medtrack. Medtrack: Biomedical Corporate Intelligence Database. Available online: http://www.medtrack.com/ (accessed on 7 February 2012).
- Thomson Reuters. Thomson Reuters Pharma Database. New York, NY, USA, 2011. Available online: https://www.thomson-pharma.com/ (accessed on 7 February 2012).
- Sawyers, C.L.; Hochhaus, A.; Feldman, E.; Goldman, J.M.; Miller, C.B.; Ottmann, O.G.; Schiffer, C.A.; Talpaz, M.; Guilhot, F.; Deininger, M.W.; et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: Results of a phase II study. Blood 2002, 99, 3530–3539. [Google Scholar]
- Kantarjian, H.M.; Cortes, J.; O’Brien, S.; Giles, F.J.; Albitar, M.; Rios, M.B.; Shan, J.; Faderl, S.; Garcia-Manero, G.; Thomas, D.A.; et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood 2002, 99, 3547–3553. [Google Scholar]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Hochhaus, A.; O’Brien, S.G.; Guilhot, F.; Druker, B.J.; Branford, S.; Foroni, L.; Goldman, J.M.; Müller, M.C.; Radich, J.P.; Rudoltz, M.; et al. IRIS Investigators. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23, 1054–1061. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevärn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Björeman, M.; Flogegård, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef]
- Meng, J.; Dai, B.; Fang, B.; Bekele, B.N.; Bornmann, W.G.; Sun, D.; Peng, Z.; Herbst, R.S.; Papadimitrakopoulou, V.; Minna, J.D.; et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLOS One 2010, 5. [Google Scholar] [CrossRef]
- Novartis. Novartis Oncology Products. Available online: http://www.novartisoncology.com/novartis-oncology-products/ (accessed on 7 February 2012).
- Engelman, J.A.; Jänne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar] [CrossRef]
- Marchant, J. Key Trends in Drug-Diagnostic Co-Development; Business Insight Ltd.: Dallas, TX, USA, 2009. [Google Scholar]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.; Audeh, M.W.; Weitze, J.N.; Friedlander, M.; Carmichael, J. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J. Clin. Oncol. ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2009, 27(15S), CRA501. [Google Scholar]
- Audeh, M.W.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Carmichael, J.; Tutt, A. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. J. Clin. Oncol. ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2009, 27 (15S), 5500. [Google Scholar]
- Sessa, C.; Guibal, A.; Del Conte, G.; Rüegg, C. Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: Tools or decorations? Nat. Clin. Pract. Oncol. 2008, 5, 378–391. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Bhutia, S.K.; Mallick, S.K.; Maiti, T.K. Tumour escape mechanisms and their therapeutic implications in combination tumour therapy. Cell Biol. Int. 2010, 34, 553–563. [Google Scholar] [CrossRef]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O’Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef]
- Carlson, R.H. Ponatinib effective in heavily pretreated CML patients with T3151 mutation. Oncol. Times UK 2011, 8, 17–18. [Google Scholar]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. Herceptin: Mechanisms of action and resistance. Cancer Lett. 2006, 232, 123–138. [Google Scholar] [CrossRef]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Sussman, L. Matching targeted therapies to tumor’s specific gene mutations key to personalized cancer treatment. Eurekalert. Available online: http://www.eurekalert.org/pub_releases/2011-06/uotm-mtt060311.php (accessed on 20 January 2012).
- Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B. Future of personalized medicine in oncology: A systems biology approach. J. Clin. Oncol. 2010, 28, 2777–2783. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fleck, R.; Bach, D. Trends in Personalized Therapies in Oncology: The (Venture) Capitalist’s Perspective. J. Pers. Med. 2012, 2, 15-34. https://doi.org/10.3390/jpm2010015
Fleck R, Bach D. Trends in Personalized Therapies in Oncology: The (Venture) Capitalist’s Perspective. Journal of Personalized Medicine. 2012; 2(1):15-34. https://doi.org/10.3390/jpm2010015
Chicago/Turabian StyleFleck, Roman, and Daniel Bach. 2012. "Trends in Personalized Therapies in Oncology: The (Venture) Capitalist’s Perspective" Journal of Personalized Medicine 2, no. 1: 15-34. https://doi.org/10.3390/jpm2010015
APA StyleFleck, R., & Bach, D. (2012). Trends in Personalized Therapies in Oncology: The (Venture) Capitalist’s Perspective. Journal of Personalized Medicine, 2(1), 15-34. https://doi.org/10.3390/jpm2010015