
Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay–Sachs Disease
 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Materials & Methods
2.1. Experimental Animals and Cell Culture
2.2. Recombinant β-Hexosaminidases a Production
2.3. rhHex-A Treatment
2.4. q-RT PCR Analysis
2.5. Immunocytochemistry Analysis
2.6. Statistical Analysis
3. Results
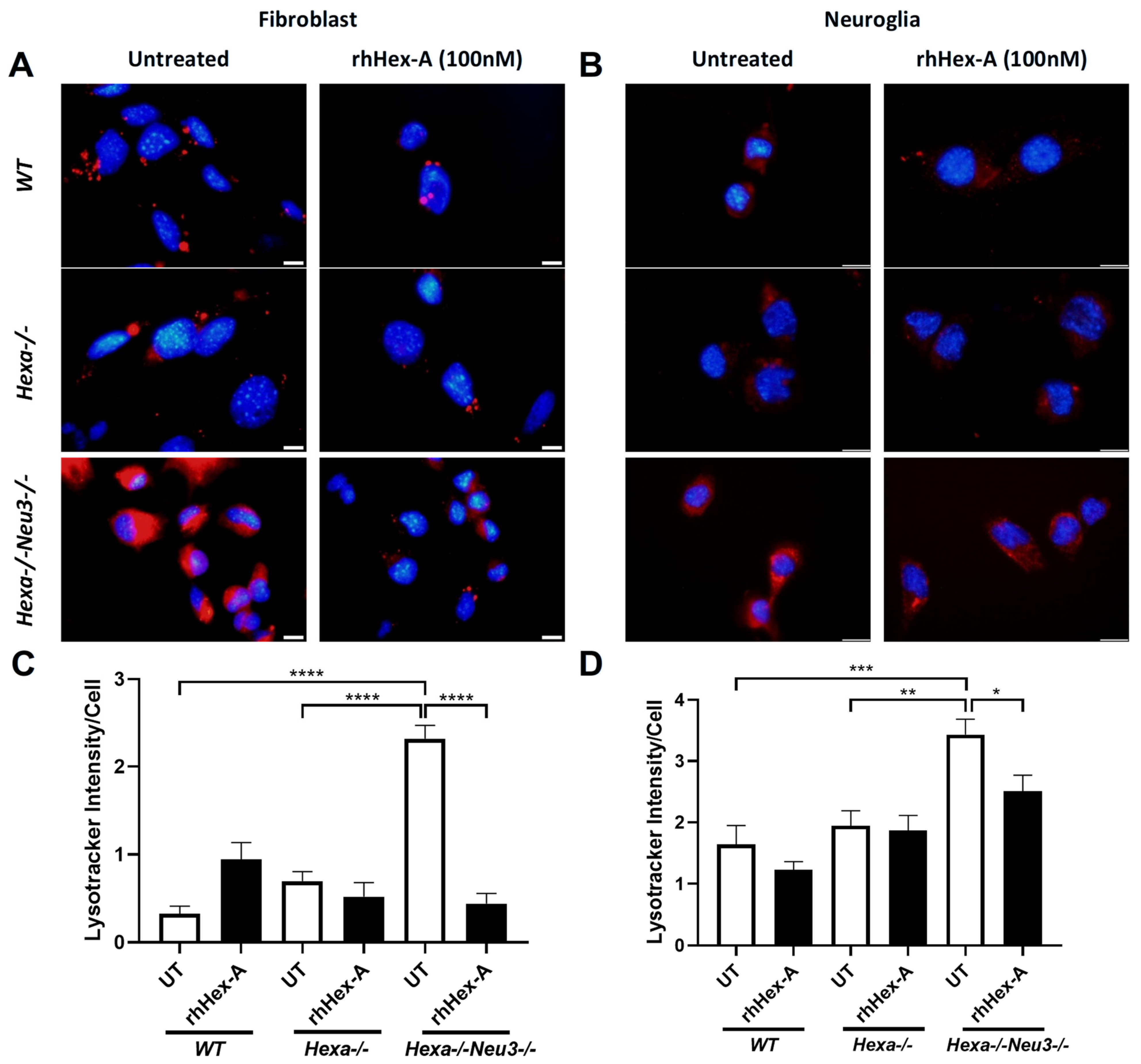
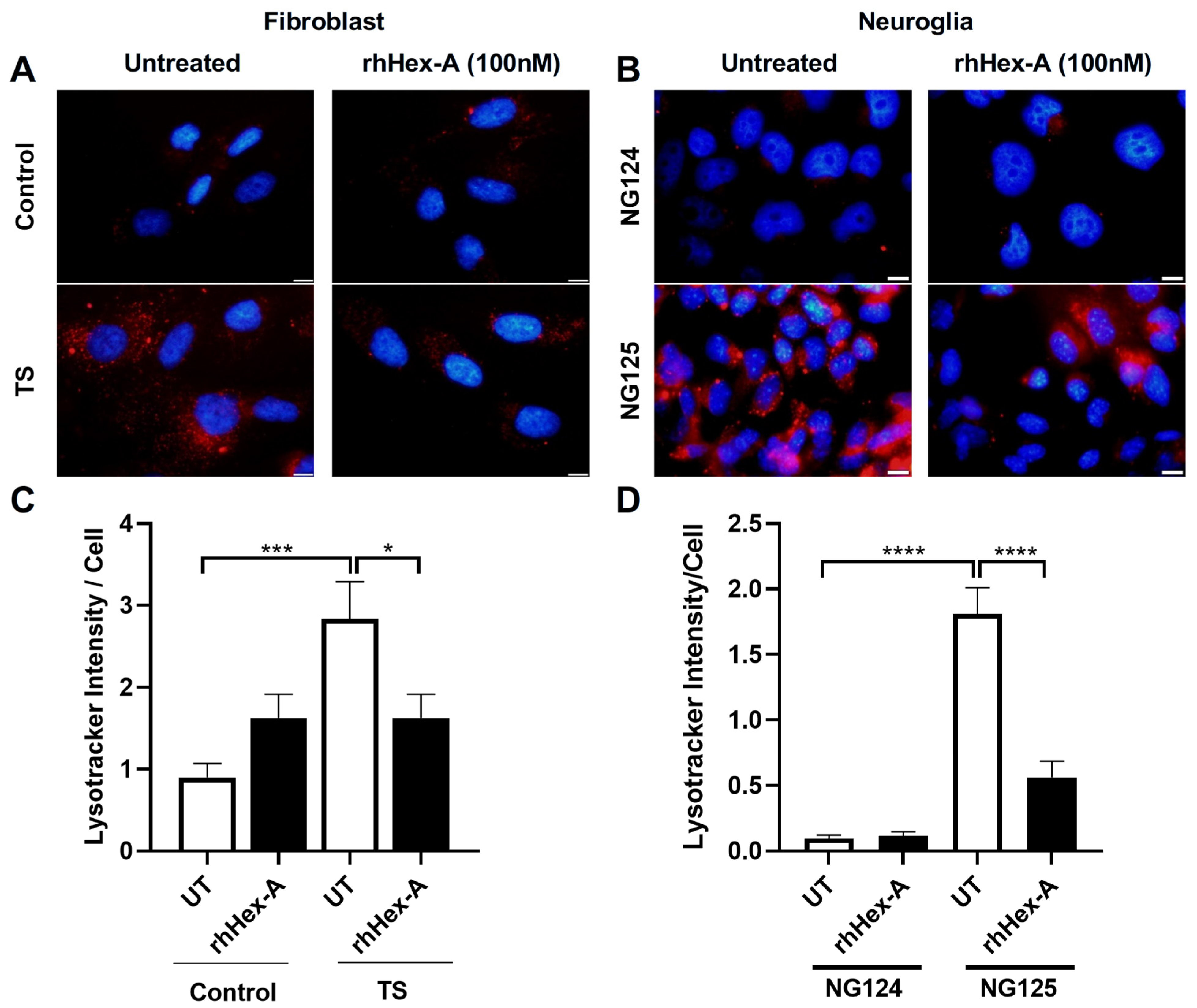
3.1. Human Recombinant Hex-A (rhHex-A) Treatment Significantly Reduces Lysosomal Mass in TS Cells
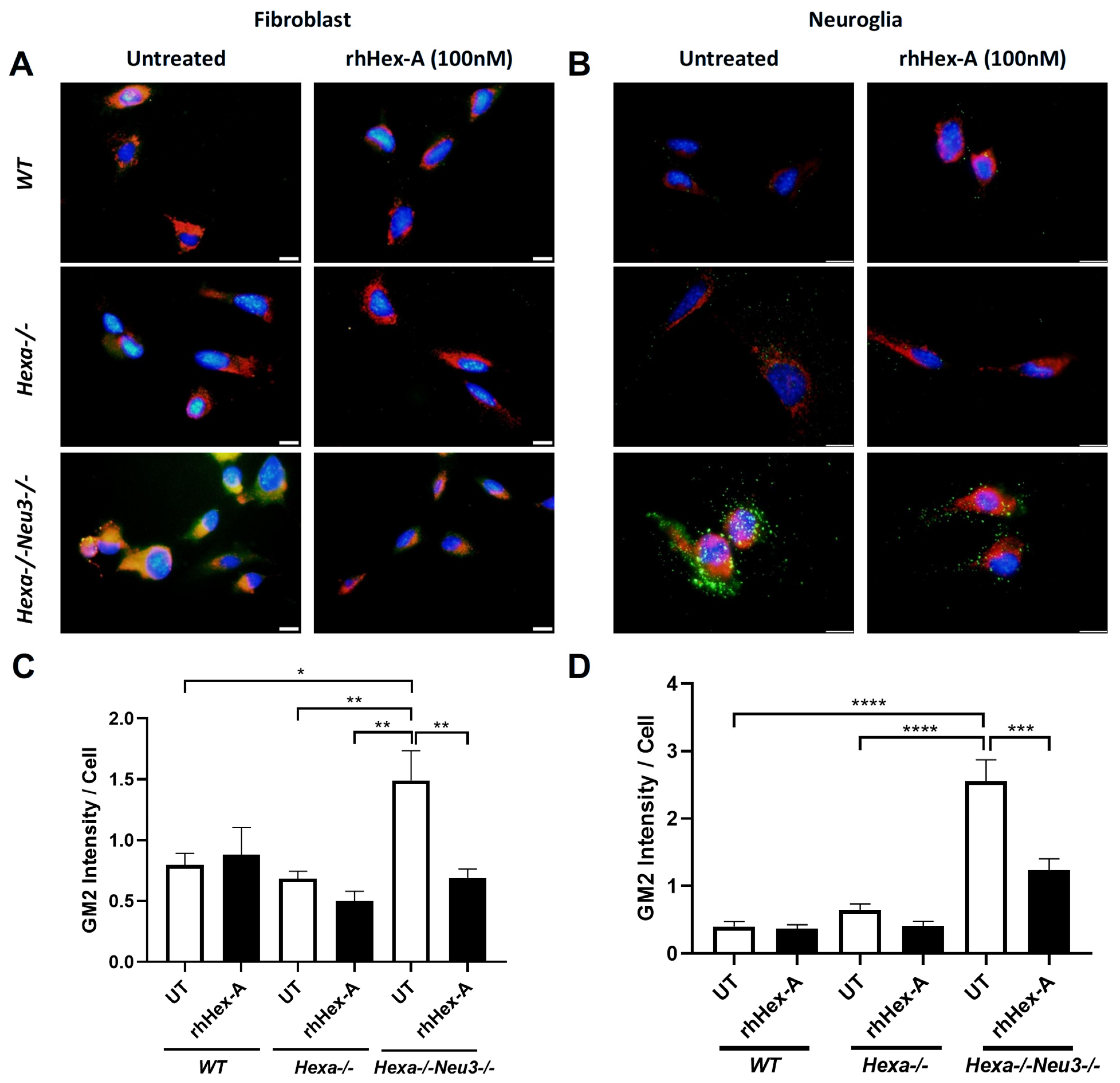
3.2. Abnormal GM2 Accumulation Was Mitigated by rhHex-A Treatment in TS Cells
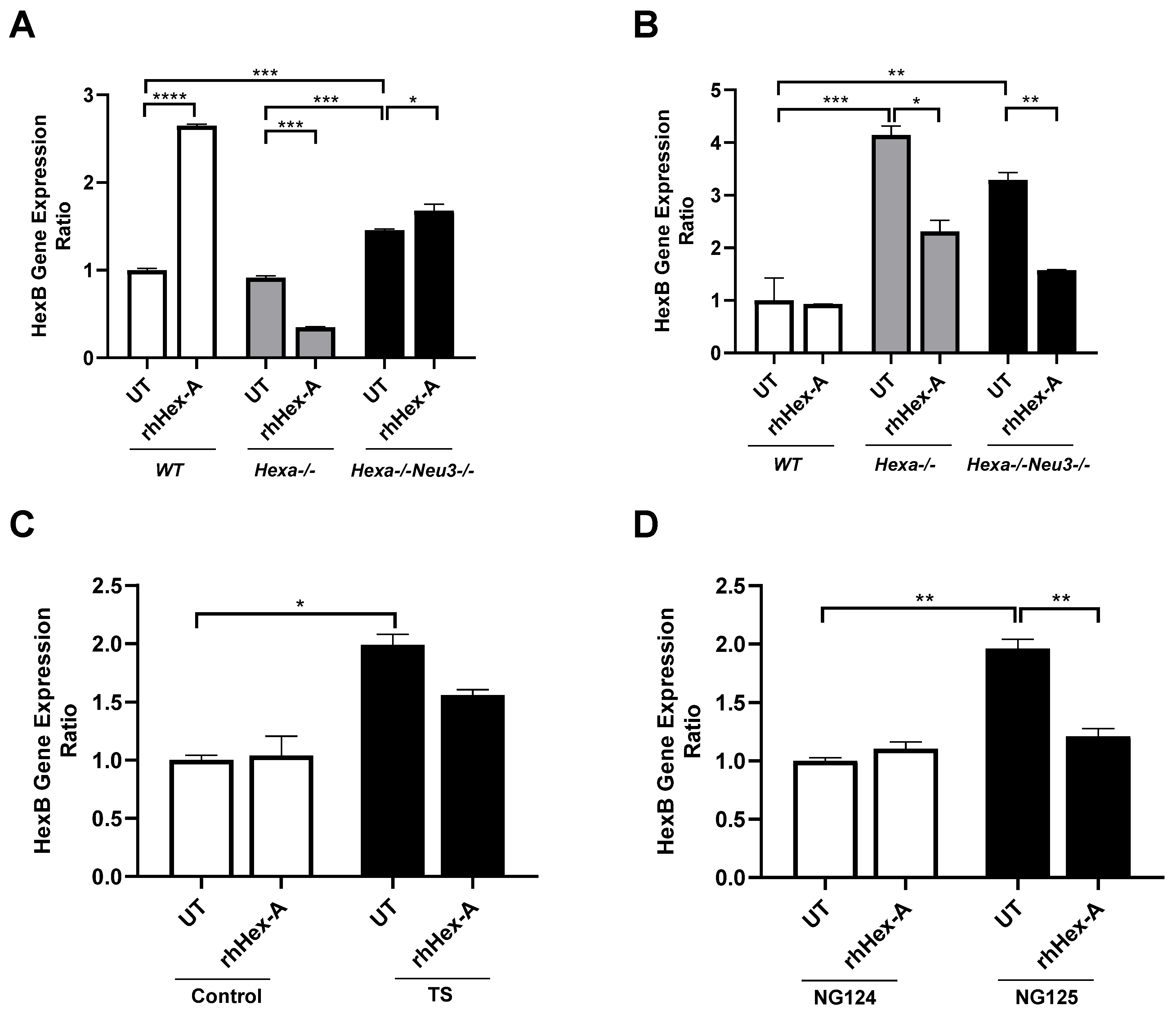
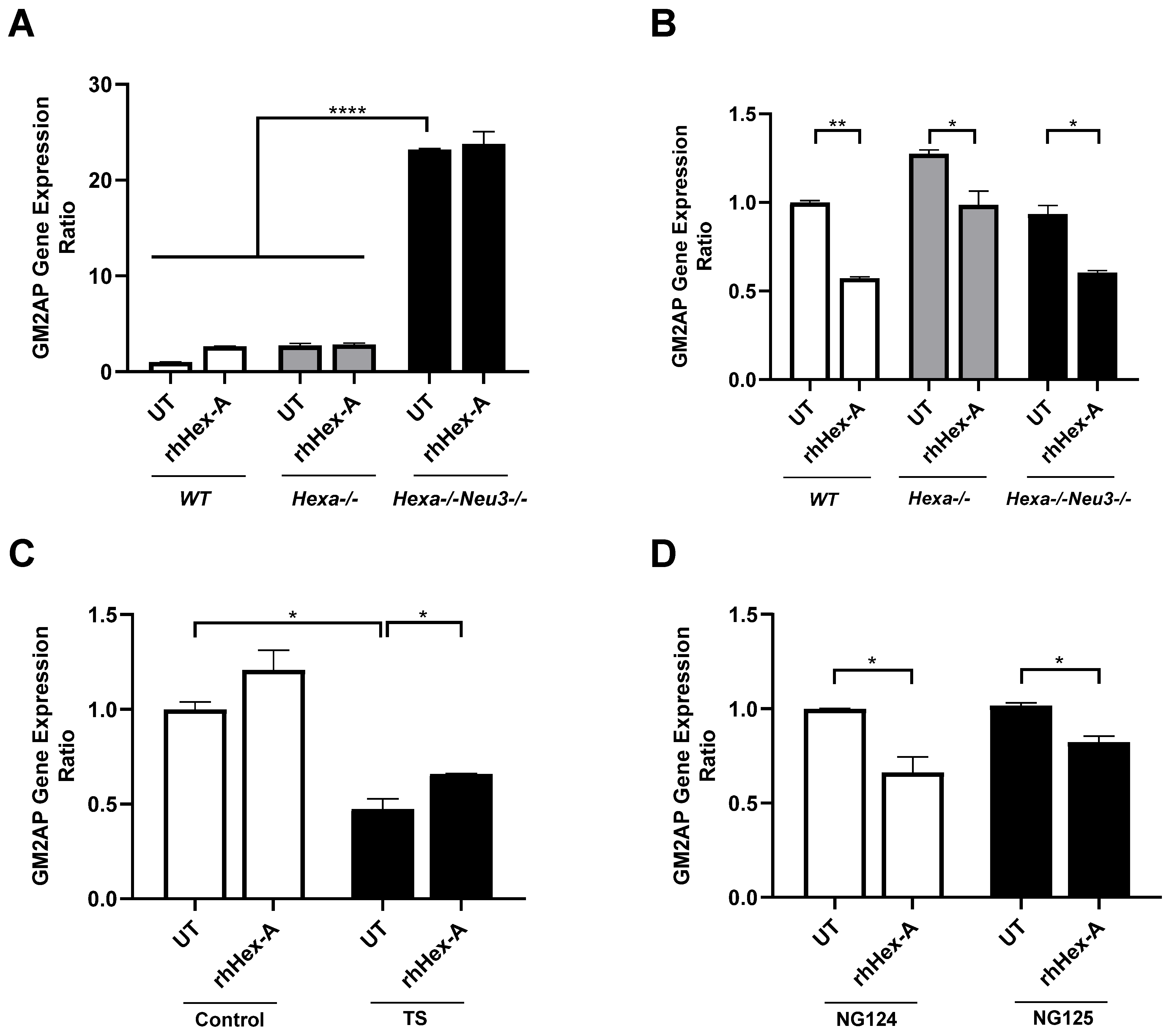
3.3. The Elevated Expression Level of HexB Gene After rhHex-A Treatment in Hexa-/-Neu3-/- Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Lysosomal Glycosphingolipid Storage Diseases. Annu. Rev. Biochem. 2019, 88, 461–485. [Google Scholar] [CrossRef]
- Beck, M.; Clarke, J.T.R.; Sandhoff, K. The Gangliosidoses. In Lysosomal Storage Disorders; Mehta, A.B., Winchester, B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar] [CrossRef]
- Yuziuk, J.A.; Bertoni, C.; Beccari, T.; Orlacchio, A.; Wu, Y.Y.; Li, S.C.; Li, Y.T. Specificity of mouse GM2 activator protein and beta-N-acetylhexosaminidases A and B. Similarities and differences with their human counterparts in the catabolism of GM2. J. Biol. Chem. 1998, 273, 66–72. [Google Scholar] [CrossRef]
- Seyrantepe, V.; Demir, S.A.; Timur, Z.K.; Von Gerichten, J.; Marsching, C.; Erdemli, E.; Oztas, E.; Takahashi, K.; Yamaguchi, K.; Ates, N.; et al. Murine Sialidase Neu3 facilitates GM2 degradation and bypass in mouse models of Tay-Sachs disease. Exp. Neurol. 2018, 299 Pt A, 26–41. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genom. Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Tamura, T.; Tsuji, D.; Dohzono, Y.; Kitakaze, K.; Ohno, K.; Saito, S.; Sakuraba, H.; Itoh, K. Therapeutic potential of intracerebroventricular replacement of modified human β-hexosaminidase B for GM2 gangliosidosis. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Espejo Mojica, A.J.; Mosquera, A.; Rodríguez-López, A.; Díaz, D.; Beltrán, L.; Hernández, F.L.; Alméciga Díaz, C.J.; Barrera, L.A. Characterization of recombinant human lysosomal beta-hexosaminidases produced in the methylotrophic yeast Pichia pastoris. Univ. Sci. 2016, 21, 195–217. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef]
- Espejo-Mojica, A.J.; Rodríguez-López, A.; Li, R.; Zheng, W.; Alméciga-Díaz, C.J.; Dulcey-Sepúlveda, C.; Combariza, G.; Barrera, L.A. Human recombinant lysosomal β-Hexosaminidases produced in Pichia pastoris efficiently reduced lipid accumulation in Tay-Sachs fibroblasts. Am. J. Med. genetics Part C Semin. Med. Genet. 2020, 184, 885–895. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Amsterdam, D.; Schneck, L. GM2 ganglioside in fetal Tay-Sachs disease brain cultures: A model system for the disease. Brain Res. 1976, 111, 109–117. [Google Scholar] [CrossRef]
- Fernandes, M.J.; Yew, S.; Leclerc, D.; Henrissat, B.; Vorgias, C.E.; Gravel, R.A.; Hechtman, P.; Kaplan, F. Identification of candidate active site residues in lysosomal beta-hexosaminidase A. J. Biol. Chem. 1997, 272, 814–820. [Google Scholar] [CrossRef]
- Shapira, E.; Blitzer, M.G.; Miller, J.B.; Africk, D.K. Biochemical Genetics: A Laboratory Manual; N.Y.O.U. Press: New York, NY, USA; Oxford, UK, 1989. [Google Scholar]
- Rodríguez-López, A.; Alméciga-Díaz, C.J.; Sánchez, J.; Moreno, J.; Beltran, L.; Díaz, D.; Pardo, A.; Ramírez, A.M.; Espejo-Mojica, A.J.; Pimentel, L.; et al. Recombinant human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) was produced in the methylotrophic yeast Pichia pastoris. Sci. Rep. 2016, 6, 29329. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, A.; Pimentel-Vera, L.N.; Espejo-Mojica, A.J.; Van Hecke, A.; Tiels, P.; Tomatsu, S.; Callewaert, N.; Alméciga-Díaz, C.J. Characterization of Human Recombinant N-Acetylgalactosamine-6-Sulfate Sulfatase Produced in Pichia pastoris as a Potential Enzyme for Mucopolysaccharidosis IVA Treatment. J. Pharm. Sci. 2019, 108, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, N.; Rodríguez-Lopez, A.; Díaz, S.; Losada, J.C.; Díaz-Rincón, D.J.; Cardona, C.; Espejo-Mojica, Á.J.; Ramírez, A.M.; Ruiz, F.; Landázuri, P.; et al. Production and characterization of a human lysosomal recombinant iduronate-2-sulfatase produced in Pichia pastoris. Biotechnol. Appl. Biochem. 2018, 65, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Begley, D.J.; Bellettato, C.M.; Scarpa, M. Central nervous system aspects, neurodegeneration, and the blood-brain barrier. In Enzyme Replacement Therapies in Neurodegenerative Disorders; Mehta, A.B., Winchester, B., Eds.; Wiley: New York, NY, USA, 2022. [Google Scholar] [CrossRef]
- Sun, Y.; Liou, B.; Chu, Z.; Fannin, V.; Blackwood, R.; Peng, Y.; Grabowski, G.A.; Davis, H.W.; Qi, X. Systemic enzyme delivery by blood-brain barrier-penetrating SapC-DOPS nanovesicles for treatment of neuronopathic Gaucher disease. EBioMedicine 2020, 55, 102735. [Google Scholar] [CrossRef]
- Fellgiebel, A.; Gartenschläger, M.; Wildberger, K.; Scheurich, A.; Desnick, R.J.; Sims, K. Enzyme replacement therapy stabilized white matter lesion progression in Fabry disease. Cerebrovasc. Dis. 2014, 38, 448–456. [Google Scholar] [CrossRef]
- Chien, Y.H.; Tsai, W.H.; Chang, C.L.; Chiu, P.C.; Chou, Y.Y.; Tsai, F.J.; Wong, S.L.; Lee, N.C.; Hwu, W.L. Earlier and higher dosing of alglucosidase alfa improves outcomes in patients with infantile-onset Pompe disease: Evidence from real-world experiences. Mol. Genet. Metab. Rep. 2020, 23, 100591. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef]
- de Los Reyes, E.; Lehwald, L.; Augustine, E.F.; Berry-Kravis, E.; Butler, K.; Cormier, N.; Demarest, S.; Lu, S.; Madden, J.; Olaya, J.; et al. Intracerebroventricular Cerliponase Alfa for Neuronal Ceroid Lipofuscinosis Type 2 Disease: Clinical Practice Considerations From US Clinics. Pediatr. Neurol. 2020, 110, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Muschol, N.; Koehn, A.; von Cossel, K.; Okur, I.; Ezgu, F.; Harmatz, P.; de Castro Lopez, M.J.; Couce, M.L.; Lin, S.P.; Batzios, S.; et al. A phase I/II study on intracerebroventricular tralesinidase alfa in patients with Sanfilippo syndrome type B. J. Clin. Investig. 2023, 133, e165076. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, M.; Kotani, M.; Tajima, Y.; Tsuji, D.; Ishibashi, Y.; Kuroki, A.; Itoh, K.; Watabe, K.; Sango, K.; Yamanaka, S.; et al. Establishment of immortalized Schwann cells from Sandhoff mice and corrective effect of recombinant human beta-hexosaminidase A on the accumulated GM2 ganglioside. J. Hum. Genet. 2005, 50, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Pennybacker, M.; Liessem, B.; Moczall, H.; Tifft, C.J.; Sandhoff, K.; Proia, R.L. Identification of Domains in Human Beta-Hexosaminidase That Determine Substrate Specificity. J. Biol. Chem. 1996, 271, 17377–17382. [Google Scholar] [CrossRef]
- Akeboshi, H.; Chiba, Y.; Kasahara, Y.; Takashiba, M.; Takaoka, Y.; Ohsawa, M.; Tajima, Y.; Kawashima, I.; Tsuji, D.; Itoh, K.; et al. Production of recombinant beta-hexosaminidase A, a potential therapeutic enzyme for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta. Appl. Environ. Microbiol. 2007, 73, 4805–4812. [Google Scholar] [CrossRef]
- Akeboshi, H.; Kasahara, Y.; Tsuji, D.; Itoh, K.; Sakuraba, H.; Chiba, Y.; Jigami, Y. Production of human beta-hexosaminidase A with highly phosphorylated N-glycans by the overexpression of the Ogataea minuta MNN4 gene. Glycobiology 2009, 19, 1002–1009. [Google Scholar] [CrossRef]
- Duarte, V.M.; Tibavija, S.S.; Rojase, H.Y.T.; Leal, A.F.; Zamora-Moreno, S.; Bojaca, J.A.; Alméciga-Díaz, C.J.; Espejo, A.J. Evaluation of hydrolytic activity of two recombinant N-acetylglucosaminidases as potential therapeutic tools for mucopolysaccharidosis type IIIB. Mol. Genet. Metab. 2023, 138, 107087. [Google Scholar] [CrossRef]
- Regier, D.S.; Proia, R.L.; D’azzo, A.; Tifft, C.J. The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy. Pediatr. Endocrinol. Rev. PER 2016, 13 (Suppl. S1), 663–673. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mHexB | F: 5′-AGTGCGAGTCCTTCCCTAGT-3′ R: 5′-ATCCGGACATCGTTTGGTGT-3′ | 412 bp |
| mGM2AP | F: 5′-GCTGGCTTCTGGGTCAAGAT-3′ R: 5′-GCACTGTGAAGTTGCTCGTG-3′ | 193 bp |
| mGAPDH | F: 5′-CCCCTTCATTGACCTCAACTAC-3′ R: 5′-ATGCATTGCTGACAATCTTGAG-3′ | 347 bp |
| hHexB | F: 5′-GTTTTGGATATTATTGCAACCATAAA-3′ R: 5′-AGTACAGATTGCTGTGGCCT-3′ | 338 bp |
| hGM2AP | F: 5′-TACCTATGGGCTTCCTTGCCAC-3′ R: 5′-GACGCTCTCTATGCGGTAGTTC-3′ | 130 bp |
| hGAPDH | F: 5′-CCCCTTCATTGACCTCAACTAC-3′ R: 5′-ATGCATTGCTGACAATCTTGAG-3′ | 347 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inci, O.K.; Leal, A.F.; Ates, N.; Súarez, D.A.; Espejo-Mojica, A.J.; Alméciga-Diaz, C.J.; Seyrantepe, V. Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay–Sachs Disease. J. Pers. Med. 2025, 15, 196. https://doi.org/10.3390/jpm15050196
Inci OK, Leal AF, Ates N, Súarez DA, Espejo-Mojica AJ, Alméciga-Diaz CJ, Seyrantepe V. Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay–Sachs Disease. Journal of Personalized Medicine. 2025; 15(5):196. https://doi.org/10.3390/jpm15050196
Chicago/Turabian StyleInci, Orhan Kerim, Andrés Felipe Leal, Nurselin Ates, Diego A. Súarez, Angela Johana Espejo-Mojica, Carlos Javier Alméciga-Diaz, and Volkan Seyrantepe. 2025. "Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay–Sachs Disease" Journal of Personalized Medicine 15, no. 5: 196. https://doi.org/10.3390/jpm15050196
APA StyleInci, O. K., Leal, A. F., Ates, N., Súarez, D. A., Espejo-Mojica, A. J., Alméciga-Diaz, C. J., & Seyrantepe, V. (2025). Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay–Sachs Disease. Journal of Personalized Medicine, 15(5), 196. https://doi.org/10.3390/jpm15050196