
Inhibitors of Immune Checkpoints: Small Molecule- and Peptide-Based Approaches
 , , and
, , and 
Abstract
1. Introduction
2. Small Molecules as Immune Checkpoint Inhibitors
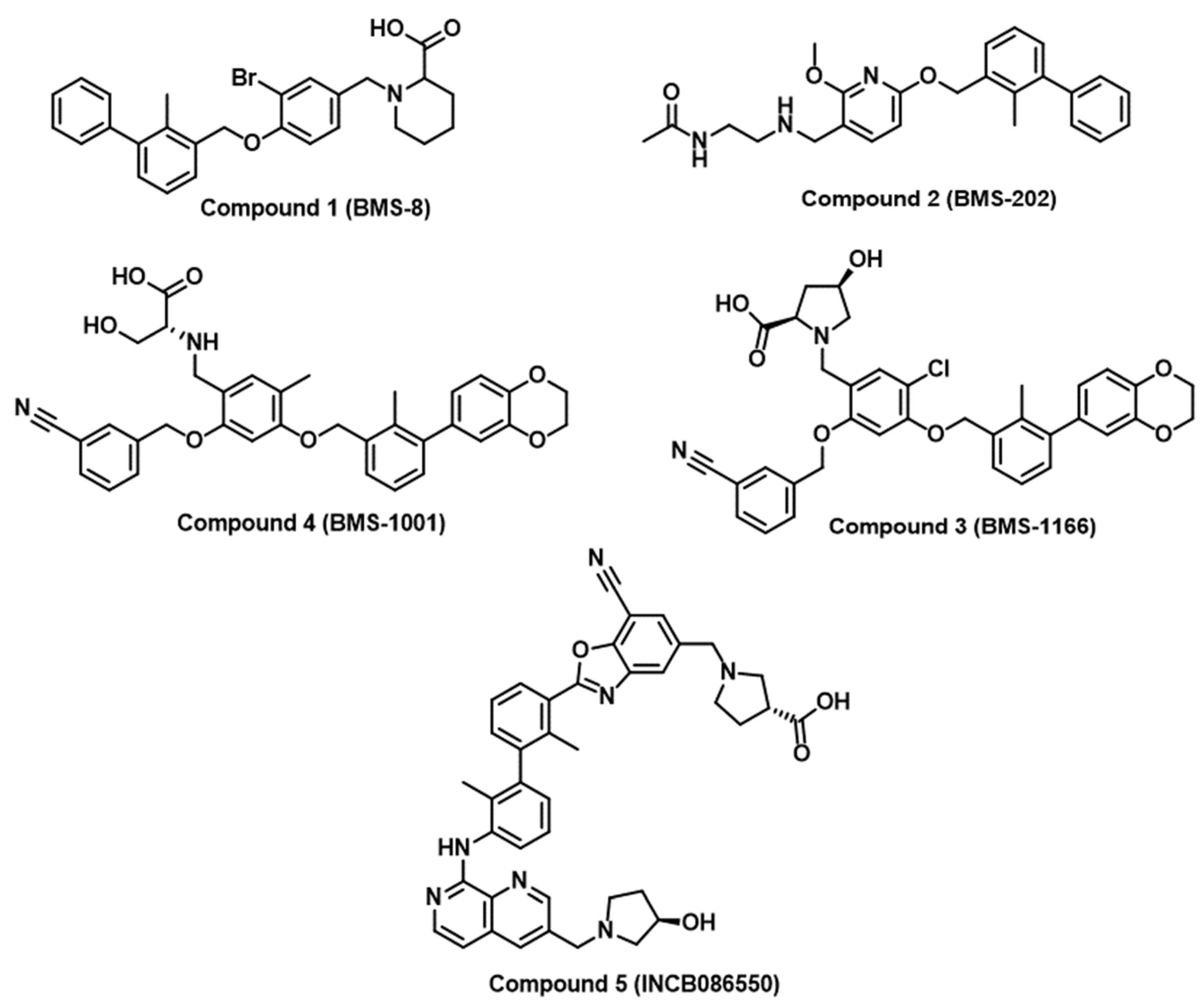
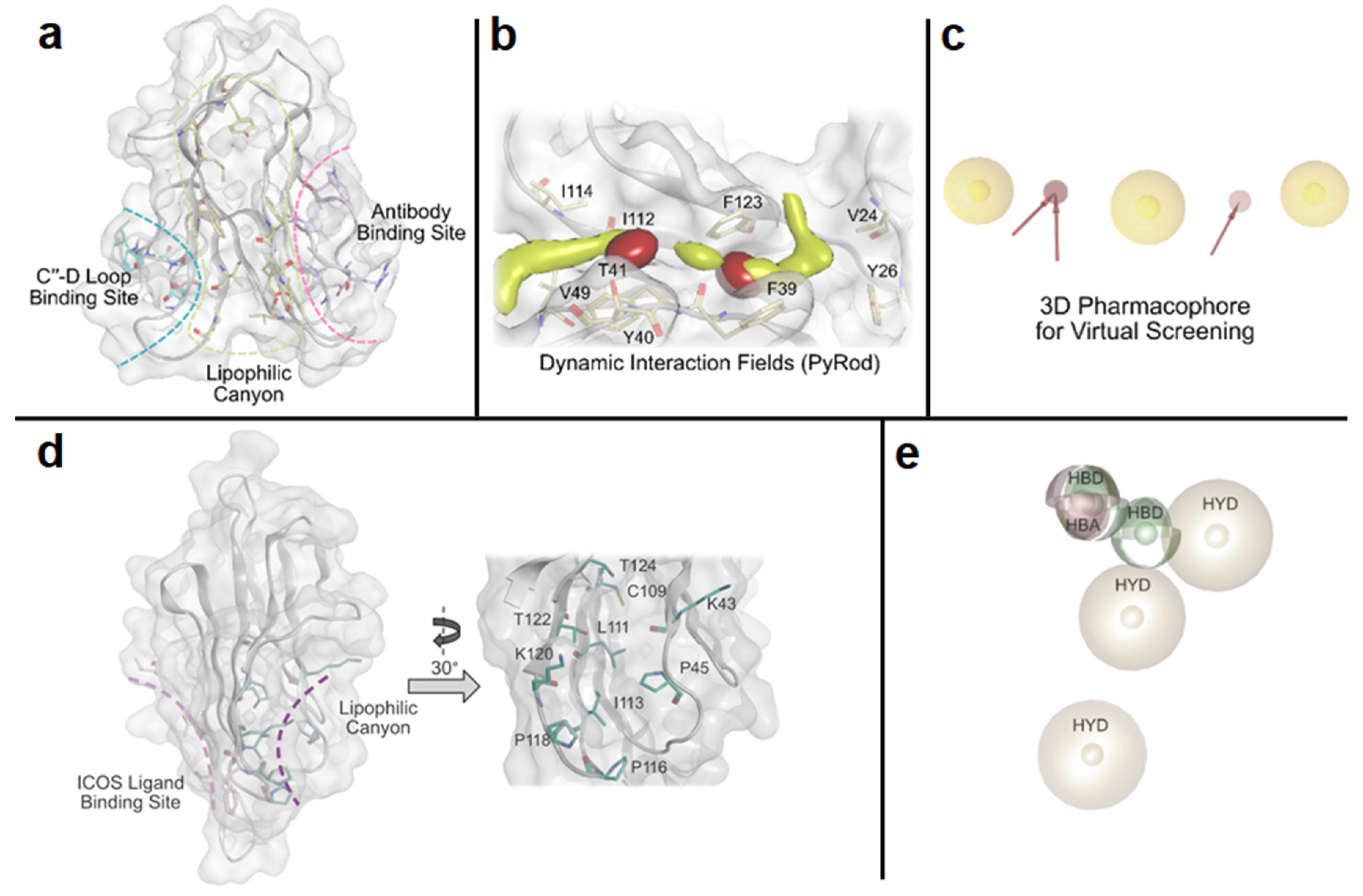
2.1. Random and Focused Screening Approaches
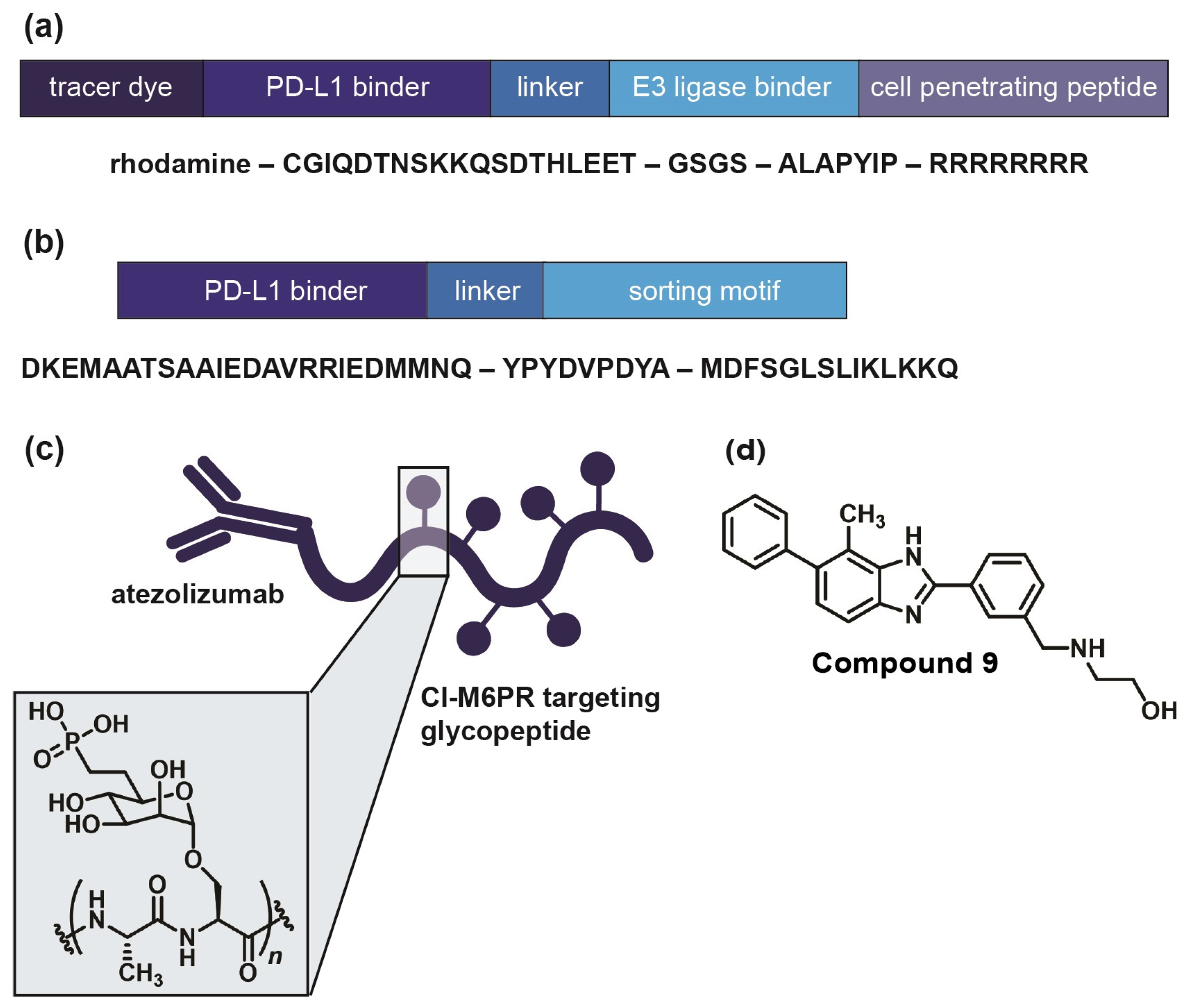
2.2. Immune Checkpoint-Targeting Degraders and Covalent Inhibitors

2.3. DNA-Encoded Library Screening as a Powerful High-Throughput Technology in Drug Discovery
3. Peptides as Immune Checkpoint Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Peptide Sequence | Method of Identification | KD (μM) | Target |
|---|---|---|---|---|
| HVEM(14–39) |  | rational design [134] | 0.102 | BTLA |
| P16 | cyc(EIDTVLTPTGWVAKRYS) | rational design [135] | 31 | CTLA-4 |
| PD-L1Pep-1 | CLQKTPKQC | phage display [137] | 0.373 | PD-L1 |
| PD-L1Pep-2 | CVRARTR | phage display [137] | 0.281 | PD-L1 |
| CLP002 | WHRSYYTWNLNT | phage display [139] | 0.366 | PD-L1 |
| LC4 | WGHSHFSHWKGR | phage display [140] | 6.86 | CTLA-4 |
| DTBP-3 | GGYTFHWHRLNP | phage display [141] | 5.60 | TIGIT |
| D4-2 | RYSAVYSIHPSW | RaPID [145] | 0.008 | CD47 |
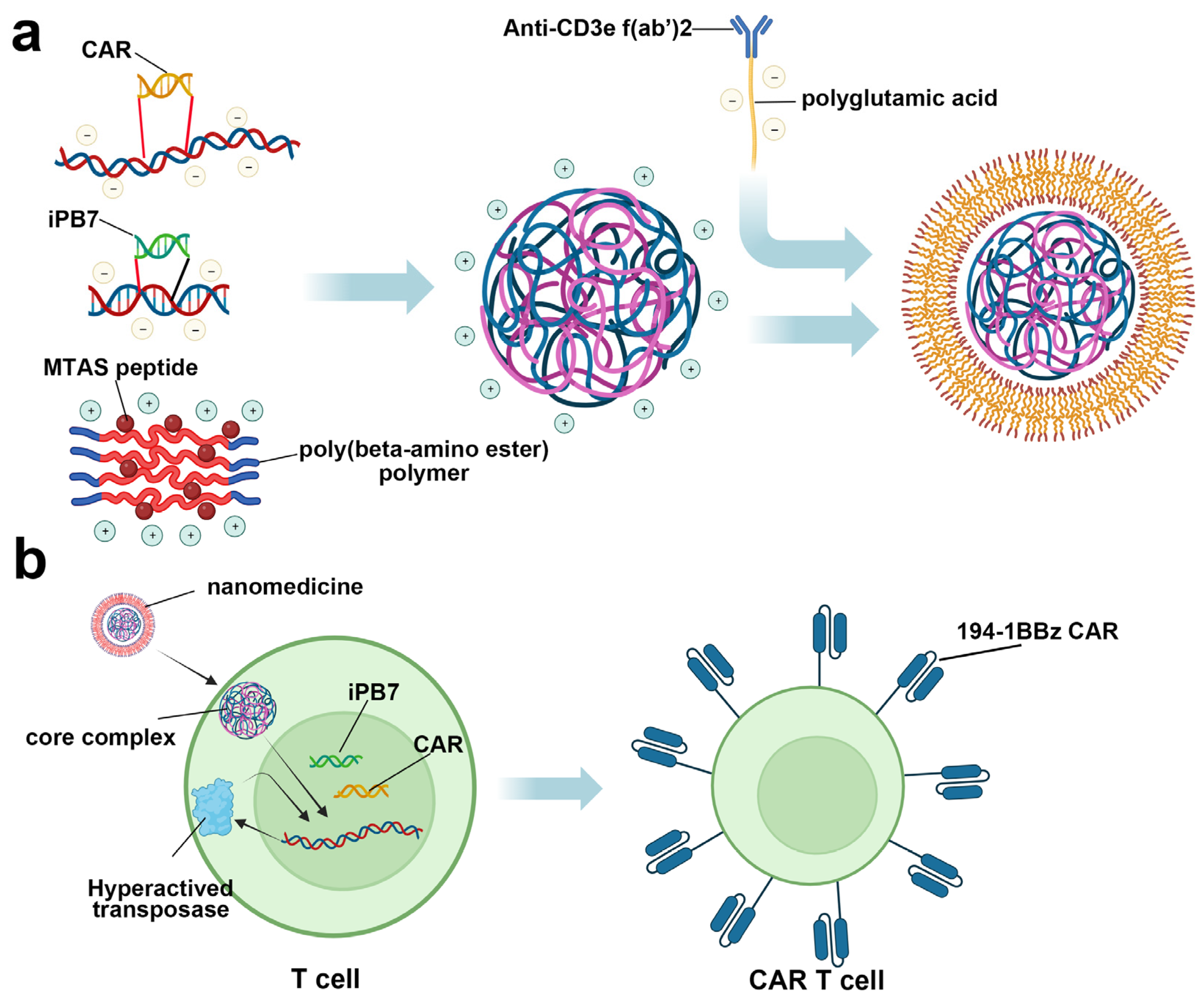
4. Nanomaterials for Cancer Immunotherapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, J. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar] [PubMed]
- Wang, Y.; Yang, S.; Wan, L.; Ling, W.; Chen, H.; Wang, J. New developments in the mechanism and application of immune checkpoint inhibitors in cancer therapy (Review). Int. J. Oncol. 2023, 63, 86. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Brábek, J. Cancer, checkpoint inhibitors, and confusion. Lancet Oncol. 2017, 18, e632. [Google Scholar] [CrossRef][Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Sathyanarayanan, V.; Neelapu, S.S. Cancer immunotherapy: Strategies for personalization and combinatorial approaches. Mol. Oncol. 2015, 9, 2043–2053. [Google Scholar] [CrossRef]
- Durant, J.R. Immunotherapy of cancer: The end of the beginning? N. Engl. J. Med. 1987, 316, 939–941. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- June, C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Investig. 2007, 117, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.A.; Schenk, A.D.; Miller, H.E.; Guleria, I.; Barth, R.J.; Huang, Y.H.; et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br. J. Haematol. 2013, 162, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Piconese, S. Regulatory-T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; June, C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; June, C.H. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J. Clin. Investig. 2010, 120, 76–80. [Google Scholar] [CrossRef]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014, 6, 1265–1278. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, L.; Zuo, Y.; Qian, H.; Liu, C. Immune Checkpoint Blockade in Cancer Immunotherapy: Mechanisms, Clinical Outcomes, and Safety Profiles of PD-1/PD-L1 Inhibitors. Arch. Immunol. Ther. Exp. 2020, 68, 36. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kroemer, G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. 2012, 1, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017, 130, 267–270. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Reilly, R.M.; Sandhu, J.; Alvarez-Diez, T.M.; Gallinger, S.; Kirsh, J.; Stern, H. Problems of delivery of monoclonal antibodies. Pharmaceutical and pharmacokinetic solutions. Clin. Pharmacokinet. 1995, 28, 126–142. [Google Scholar] [CrossRef]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef]
- Taylor, J.; Gandhi, A.; Gray, E.; Zaenker, P. Checkpoint inhibitor immune-related adverse events: A focused review on autoantibodies and B cells as biomarkers, advancements and future possibilities. Front. Immunol. 2023, 13, 991433. [Google Scholar] [CrossRef]
- Mosch, R.; Guchelaar, H.J. Immunogenicity of Monoclonal Antibodies and the Potential Use of HLA Haplotypes to Predict Vulnerable Patients. Front. Immunol. 2022, 13, 885672. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, H. Cancer Immunotherapy: Selected Targets and Small-Molecule Modulators. ChemMedChem 2016, 11, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.M.; Purvis, I.J.; Bomstad, C.N.; Labak, C.M.; Velpula, K.K.; Tsung, A.J.; Regan, J.N.; Venkataraman, S.; Vibhakar, R.; Asuthkar, S. Therapeutic targeting of immune checkpoints with small molecule inhibitors. Am. J. Transl. Res. 2019, 11, 529–541. [Google Scholar] [PubMed]
- Sasikumar, P.G.; Ramachandra, M. Small Molecule Agents Targeting PD-1 Checkpoint Pathway for Cancer Immunotherapy: Mechanisms of Action and Other Considerations for Their Advanced Development. Front. Immunol. 2022, 13, 752065. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S.; et al. Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef]
- Kerr, W.G.; Chisholm, J.D. The Next Generation of Immunotherapy for Cancer: Small Molecules Could Make Big Waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef]
- Liu, B.; Li, S.; Hu, J. Technological advances in high-throughput screening. Am. J. Pharmacogenomics 2004, 4, 263–276. [Google Scholar] [CrossRef]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Pereira, D.A.; Williams, J.A. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef]
- Koblish, H.K.; Wu, L.; Wang, L.S.; Liu, P.C.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N.; et al. Characterization of INCB086550: A Potent and Novel Small-Molecule PD-L1 Inhibitor. Cancer Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, Y.; Yu, C.; Du, H.; Liu, J.; Li, H.; Huang, S.; Zhu, Q.; Xu, Y.; Zou, Y. Discovery of Novel Small-Molecule Inhibitors of PD-1/PD-L1 Interaction via Structural Simplification Strategy. Molecules 2021, 26, 3347. [Google Scholar] [CrossRef]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, S.; Plewka, J.; Wu, C.; Zhu, M.; Yu, Q.; Musielak, B.; Wang, X.; Awadasseid, A.; Magiera-Mularz, K.; et al. Design, Synthesis, and Antitumor Activity Evaluation of 2-Arylmethoxy-4-(2,2’-dihalogen-substituted biphenyl-3-ylmethoxy) Benzylamine Derivatives as Potent PD-1/PD-L1 Inhibitors. J. Med. Chem. 2023, 66, 10579–10603. [Google Scholar] [CrossRef]
- Ważyńska, M.A.; Butera, R.; Requesens, M.; Plat, A.; Zarganes-Tzitzikas, T.; Neochoritis, C.G.; Plewka, J.; Skalniak, L.; Kocik-Krol, J.; Musielak, B.; et al. Design, Synthesis, and Biological Evaluation of 2-Hydroxy-4-phenylthiophene-3-carbonitrile as PD-L1 Antagonist and Its Comparison to Available Small Molecular PD-L1 Inhibitors. J. Med. Chem. 2023, 66, 9577–9591. [Google Scholar] [CrossRef]
- Butera, R.; Ważyńska, M.; Magiera-Mularz, K.; Plewka, J.; Musielak, B.; Surmiak, E.; Sala, D.; Kitel, R.; de Bruyn, M.; Nijman, H.W.; et al. Design, Synthesis, and Biological Evaluation of Imidazopyridines as PD-1/PD-L1 Antagonists. ACS Med. Chem. Lett. 2021, 12, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Acúrcio, R.C.; Pozzi, S.; Carreira, B.; Pojo, M.; Gómez-Cebrián, N.; Casimiro, S.; Fernandes, A.; Barateiro, A.; Farricha, V.; Brito, J.; et al. Therapeutic targeting of PD-1/PD-L1 blockade by novel small-molecule inhibitors recruits cytotoxic T cells into solid tumor microenvironment. J. Immunother. Cancer 2022, 10, e004695. [Google Scholar] [CrossRef] [PubMed]
- Lung, J.; Hung, M.S.; Lin, Y.C.; Hung, C.H.; Chen, C.C.; Lee, K.D.; Tsai, Y.H. Virtual Screening and In Vitro Evaluation of PD-1 Dimer Stabilizers for Uncoupling PD-1/PD-L1 Interaction from Natural Products. Molecules 2020, 25, 5293. [Google Scholar] [CrossRef] [PubMed]
- Pushkaran, A.C.; Kumaran, K.; Maria, A.; Biswas, R.; Mohan, C.G. Identification of a PD1/PD-L1 inhibitor by structure-based pharmacophore modelling, virtual screening, molecular docking and biological evaluation. Mol. Inform. 2023, 42, e2200254. [Google Scholar] [CrossRef] [PubMed]
- Tran-Nguyen, V.K.; Simeon, S.; Junaid, M.; Ballester, P.J. Structure-based virtual screening for PDL1 dimerizers: Evaluating generic scoring functions. Curr. Res. Struct. Biol. 2022, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Du, J.; Wang, H.; Chen, C.; Jiao, L.; Cheng, X.; Zhou, X.; Chen, S.; Gou, S.; Zhao, W.; et al. Repositioning liothyronine for cancer immunotherapy by blocking the interaction of immune checkpoint TIGIT/PVR. Cell Commun. Signal 2020, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Jiang, J.W.; Qie, C.X.; Xuan, C.X.; Hu, X.L.; Liu, W.M.; Chen, W.T.; Liu, J. Identification of active small-molecule modulators targeting the novel immune checkpoint VISTA. BMC Immunol. 2021, 22, 55. [Google Scholar] [CrossRef]
- Zhou, X.; Jiao, L.; Qian, Y.; Dong, Q.; Sun, Y.; Zheng, W.V.; Zhao, W.; Zhai, W.; Qiu, L.; Wu, Y.; et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules 2021, 11, 706. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.A.; Talagayev, V.; Pach, S.; Wolber, G.; Gabr, M.T. Discovery of Small-Molecule TIM-3 Inhibitors for Acute Myeloid Leukemia Using Pharmacophore-Based Virtual Screening. J. Med. Chem. 2023, 66, 11464–11475. [Google Scholar] [CrossRef]
- Calvo-Barreiro, L.; Talagayev, V.; Pach, S.; Abdel-Rahman, S.A.; Wolber, G.; Gabr, M.T. Discovery of ICOS-Targeted Small Molecules Using Pharmacophore-Based Screening. ChemMedChem 2023, 18, e202300305. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.A.; Zhang, L.; Gabr, M.T. Development of a high-throughput TR-FRET screening assay for LAG-3/FGL1 interaction. SLAS Discov. 2023, 28, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Rehman, A.U.; Gabr, M.T. Discovery of First-in-Class Small Molecule Inhibitors of Lymphocyte Activation Gene 3 (LAG-3). ACS Med. Chem. Lett. 2023, 14, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Świderek, K.; Gabr, M.T. First-in-class small molecule inhibitors of ICOS/ICOSL interaction as a novel class of immunomodulators. RSC Med. Chem. 2023, 14, 1767–1777. [Google Scholar] [CrossRef]
- Gabr, M.T.; Gambhir, S.S. Discovery and Optimization of Small-Molecule Ligands for V-Domain Ig Suppressor of T-Cell Activation (VISTA). J. Am. Chem. Soc. 2020, 142, 16194–16198. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. Engl. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Joyce, R.P.; Hu, V.W.; Wang, J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): An orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med. Chem. Res. 2022, 31, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar] [PubMed]
- Yap, T.A.; Vidal, L.; Adam, J.; Stephens, P.; Spicer, J.; Shaw, H.; Ang, J.; Temple, G.; Bell, S.; Shahidi, M.; et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, 3965–3972. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Q.; Klauser, P.C.; Li, M.; Zheng, F.; Wang, N.; Li, X.; Zhang, Q.; Fu, X.; Wang, Q.; et al. Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 2020, 182, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.Y.; Shi, Y.Y.; Wang, A.J.; Liu, X.L.; Liu, M.; Cai, H.B. High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells. Cell Death Dis. 2022, 13, 924. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Ren, Y.; Cao, H.; Chen, J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur. J. Med. Chem. 2020, 199, 112377. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Brosseau, J.P.; Shi, H. Targeted degradation of immune checkpoint proteins: Emerging strategies for cancer immunotherapy. Oncogene 2020, 39, 7106–7113. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Deng, S.; Zhou, X.; Xu, J. Checkpoints Under Traffic Control: From and to Organelles. Adv. Exp. Med. Biol. 2020, 1248, 431–453. [Google Scholar]
- Wang, H.; Yao, H.; Li, C.; Shi, H.; Lan, J.; Li, Z.; Zhang, Y.; Liang, L.; Fang, J.Y.; Xu, J. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 2019, 15, 42–50. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Prata, M.J.; Alves, S. A shortcut to the lysosome: The mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 2012, 107, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Yu, L.; Huang, X.; Wang, X.; Han, D.; Yang, Y.; Liu, Z. Covalent LYTAC Enabled by DNA Aptamers for Immune Checkpoint Degradation Therapy. J. Am. Chem. Soc. 2023, 145, 24506–24521. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S. The Autophagy Lysosomal Pathway and Neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a033993. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef]
- Endicott, S.J.; Boynton, D.N.; Beckmann, L.J.; Miller, R.A. Long-lived mice with reduced growth hormone signaling have a constitutive upregulation of hepatic chaperone-mediated autophagy. Autophagy 2021, 17, 612–625. [Google Scholar] [CrossRef]
- Wang, T.; Wang, K.; Zhang, Y.; Zhang, K.; Cai, S.; Jiang, S.; Xiao, Y.; Zhang, X. Novel Benzimidazoles as Potent Small-Molecule Inhibitors and Degraders of V-Domain Ig Suppressor of T-Cell Activation (VISTA). J. Med. Chem. 2023, 66, 11881–11892. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.X.; Mallet, W.G.; Huang, A.Y.; Maxfield, F.R. Endocytosed cation-independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol. Biol. Cell 2004, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Satz, A.L.; Brunschweiger, A.; Flanagan, M.E.; Gloger, A.; Hansen, N.J.; Kuai, L.; Kunig, V.B.; Lu, X.; Madsen, D.; Marcaurelle, L.A.; et al. DNA-encoded chemical libraries. Nat. Rev. Methods Primers 2022, 2, 3. [Google Scholar] [CrossRef]
- Brenner, S.; Lerner, R.A. Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. USA 1992, 89, 5381–5383. [Google Scholar] [CrossRef] [PubMed]
- Kunig, V.B.K.; Potowski, M.; Škopić, M.; Brunschweiger, A. Scanning Protein Surfaces with DNA-Encoded Libraries. ChemMedChem 2021, 16, 1048–1062. [Google Scholar] [CrossRef]
- Clark, M.A.; Acharya, R.A.; Arico-Muendel, C.C.; Belyanskaya, S.L.; Benjamin, D.R.; Carlson, N.R.; Centrella, P.A.; Chiu, C.H.; Creaser, S.P.; Cuozzo, J.W.; et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 2009, 5, 647–654. [Google Scholar] [CrossRef]
- Gironda-Martínez, A.; Donckele, E.J.; Samain, F.; Neri, D. DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci. 2021, 4, 1265–1279. [Google Scholar] [CrossRef]
- Fitzgerald, P.R.; Paegel, B.M. DNA-Encoded Chemistry: Drug Discovery from a Few Good Reactions. Chem. Rev. 2021, 121, 7155–7177. [Google Scholar] [CrossRef]
- Xie, J.; Wang, S.; Ma, P.; Ma, F.; Li, J.; Wang, W.; Lu, F.; Xiong, H.; Gu, Y.; Zhang, S.; et al. Selection of Small Molecules that Bind to and Activate the Insulin Receptor from a DNA-Encoded Library of Natural Products. iScience 2020, 23, 101197. [Google Scholar] [CrossRef] [PubMed]
- Favalli, N.; Bassi, G.; Pellegrino, C.; Millul, J.; De Luca, R.; Cazzamalli, S.; Yang, S.; Trenner, A.; Mozaffari, N.L.; Myburgh, R.; et al. Stereo- and regiodefined DNA-encoded chemical libraries enable efficient tumour-targeting applications. Nat. Chem. 2021, 13, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Harbut, M.B.; Kung, P.P.; Karpowich, N.K.; Branson, J.D.; Grant, J.C.; Hagan, D.; Pascual, H.A.; Bai, G.; Zavareh, R.B.; et al. Identification of small-molecule protein-protein interaction inhibitors for NKG2D. Proc. Natl. Acad. Sci. USA 2023, 120, e2216342120. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Du, C.; Xie, X. G protein-coupled receptors as therapeutic targets for multiple sclerosis. Cell Res. 2012, 22, 1108–1128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Abosamak, N.E.R.; Shahin, M.H. Beta2 Receptor Agonists and Antagonists. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ahn, S.; Kahsai, A.W.; Pani, B.; Wang, Q.T.; Zhao, S.; Wall, A.L.; Strachan, R.T.; Status, D.P.; Wingler, L.M.; Sun, L.D.; et al. Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. USA 2017, 114, 1708–1713. [Google Scholar] [CrossRef]
- Ahn, S.; Pani, B.; Kahsai, A.W.; Olsen, E.K.; Husemoen, G.; Vestergaard, M.; Jin, L.; Zhao, S.; Wingler, L.M.; Rambarat, P.K.; et al. Small-Molecule Positive Allosteric Modulators of the β2-Adrenoceptor Isolated from DNA-Encoded Libraries. Mol. Pharmacol. 2018, 94, 850–861. [Google Scholar] [CrossRef]
- Liang, Q.; He, J.; Zhao, X.; Xue, Y.; Zuo, H.; Xu, R.; Jin, Y.; Wang, J.; Li, Q.; Zhao, X. Selective Discovery of GPCR Ligands within DNA-Encoded Chemical Libraries Derived from Natural Products: A Case Study on Antagonists of Angiotensin II Type I Receptor. J. Med. Chem. 2021, 64, 4196–4205. [Google Scholar] [CrossRef]
- Liang, Q.; Zhao, X.; Fu, X.; Wang, J.; Li, Q.; Zhao, X. Identification of selective ligands targeting two GPCRs by receptor-affinity chromatography coupled with high-throughput sequencing techniques. Bioorganic Chem. 2021, 112, 104986. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Yu, M.; Xu, H.; Zhong, Z.; Li, Z.; Guo, Y.; Zhang, T.; Zeng, Z.; Jin, F.; He, X. Discovery of TIGIT inhibitors based on DEL and machine learning. Front. Chem. 2022, 10, 982539. [Google Scholar] [CrossRef]
- Manieri, N.A.; Chiang, E.Y.; Grogan, J.L. TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol. 2017, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Wang, Y.; Zou, S.; Zhao, Y.; Li, Y.; Zhang, C.; Guo, X.; Li, S. Construction of Peptide Library in Mammalian Cells by dsDNA-Based Strategy. ACS Omega 2022, 8, 1037–1046. [Google Scholar] [CrossRef]
- Huang, Y.; Meng, L.; Nie, Q.; Zhou, Y.; Chen, L.; Yang, S.; Fung, Y.M.E.; Li, X.; Huang, C.; Cao, Y.; et al. Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells. Nat. Chem. 2021, 13, 77–88. [Google Scholar] [CrossRef]
- Cai, B.; Kim, D.; Akhand, S.; Sun, Y.; Cassell, R.J.; Alpsoy, A.; Dykhuizen, E.C.; Van Rijn, R.M.; Wendt, M.K.; Krusemark, C.J. Selection of DNA-Encoded Libraries to Protein Targets within and on Living Cells. J. Am. Chem. Soc. 2019, 141, 17057–17061. [Google Scholar] [CrossRef]
- Xu, Y.; Piston, D.W.; Johnson, C.H. A bioluminescence resonance energy transfer (BRET) system: Application to interacting circadian clock proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 151–156. [Google Scholar] [CrossRef]
- Teske, K.A.; Su, W.; Corona, C.R.; Wen, J.; Deng, J.; Ping, Y.; Zhang, Z.; Zhang, Q.; Wilkinson, J.; Beck, M.T.; et al. DELs enable the development of BRET probes for target engagement studies in cells. Cell Chem. Biol. 2023, 30, 987–998. [Google Scholar] [CrossRef]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Harris, P.A.; King, B.W.; Bandyopadhyay, D.; Berger, S.B.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; Finger, J.N.; et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem. 2016, 59, 2163–2178. [Google Scholar] [CrossRef] [PubMed]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS ONE 2017, 12, e0179201. [Google Scholar] [CrossRef] [PubMed]
- Spodzieja, M.; Kuncewicz, K.; Sieradzan, A.; Karczyńska, A.; Iwaszkiewicz, J.; Cesson, V.; Węgrzyn, K.; Zhukov, I.; Maszota-Zieleniak, M.; Michielin, O.; et al. Disulfide-Linked Peptides for Blocking BTLA/HVEM Binding. Int. J. Mol. Sci. 2020, 21, 636. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, R.; Upreti, D.; Ishiguro, S.; Tamura, M.; Comer, J. Computational design of a cyclic peptide that inhibits the CTLA4 immune checkpoint. RSC Med. Chem. 2023, 14, 658–670. [Google Scholar] [CrossRef]
- Wu, C.H.; Liu, I.J.; Lu, R.M.; Wu, H.C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Khan, F.; Gunassekaran, G.R.; Yoo, J.D.; Vadevoo, S.M.; Permpoon, U.; Kim, S.H.; Kim, H.J.; Kim, I.S.; Han, H.; et al. Phage display-identified PD-L1-binding peptides reinvigorate T-cell activity and inhibit tumor progression. Biomaterials 2020, 247, 119984. [Google Scholar] [CrossRef]
- Jeon, I.S.; Yoo, J.D.; Gurung, S.; Kim, M.; Lee, C.; Park, E.J.; Park, R.W.; Lee, B.; Kim, S. Anticancer nanocage platforms for combined immunotherapy designed to harness immune checkpoints and deliver anticancer drugs. Biomaterials 2021, 270, 120685. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Z.; Zhang, L.; Li, Y.; Jain, A.; Barve, A.; Jin, W.; Liu, Y.; Fetse, J.; Cheng, K. Discovery of low-molecular weight anti-PD-L1 peptides for cancer immunotherapy. J. Immunother. Cancer 2019, 7, 270. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, S.; Yuan, B.; Wu, Y.; Gao, Y.; Wan, G.; Li, G. A Novel CTLA-4 affinity peptide for cancer immunotherapy by increasing the integrin αvβ3 targeting. Discov. Oncol. 2022, 13, 99. [Google Scholar] [CrossRef]
- Zhou, X.; Zuo, C.; Li, W.; Shi, W.; Zhou, X.; Wang, H.; Chen, S.; Du, J.; Chen, G.; Zhai, W.; et al. A Novel d-Peptide Identified by Mirror-Image Phage Display Blocks TIGIT/PVR for Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2020, 59, 15114–15118. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Passioura, T.; Suga, H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem. Commun. 2017, 53, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Suga, H. The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides. Acc. Chem. Res. 2021, 54, 3604–3617. [Google Scholar] [CrossRef] [PubMed]
- Hazama, D.; Yin, Y.; Murata, Y.; Matsuda, M.; Okamoto, T.; Tanaka, D.; Terasaka, N.; Zhao, J.; Sakamoto, M.; Kakuchi, Y.; et al. Macrocyclic Peptide-Mediated Blockade of the CD47-SIRPα Interaction as a Potential Cancer Immunotherapy. Cell Chem. Biol. 2020, 27, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.L.; Gordon, S.R.; Mayer, A.T.; McCracken, M.N.; Natarajan, A.; Ring, N.G.; Kimura, R.; Tsai, J.M.; Manglik, A.; Kruse, A.C.; et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E6506–E6514. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.J.; Bu, J.; Han, Y.; Drelich, A.J.; Nair, A.; Král, P.; Hong, S. Nanoparticle Conjugation Stabilizes and Multimerizes β-Hairpin Peptides to Effectively Target PD-1/PD-L1 β-Sheet-Rich Interfaces. J. Am. Chem. Soc. 2020, 142, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Kocik-Krol, J.; Skalniak, L.; Musielak, B.; Wisniewska, A.; Ciesiołkiewicz, A.; Berlicki, Ł.; Plewka, J.; Grudnik, P.; Stec, M.; et al. Structural and biological characterization of pAC65, a macrocyclic peptide that blocks PD-L1 with equivalent potency to the FDA-approved antibodies. Mol. Cancer 2023, 22, 150. [Google Scholar] [PubMed]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients with Previously Treated Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Huo, J.L.; Wang, Y.T.; Fu, W.J.; Lu, N.; Liu, Z.S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Guzman, G.; Reed, M.R.; Bielamowicz, K.; Koss, B.; Rodriguez, A. CAR-T Therapies in Solid Tumors: Opportunities and Challenges. Curr. Oncol. Rep. 2023, 25, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M. CAR T cell therapy for patients with solid tumours: Key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 2023, 21, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Daei Sorkhabi, A.; Mohamed Khosroshahi, L.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Gangadhar, T.C.; Vonderheide, R.H. Mitigating the toxic effects of anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2014, 11, 91–99. [Google Scholar] [CrossRef]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Wu, J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, W.; Nam, J.; Moon, J.J.; Kim, B.Y.S. Immunomodulating Nanomedicine for Cancer Therapy. Nano Lett. 2018, 18, 6655–6659. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Watkins-Schulz, R.; Junkins, R.D.; David, C.N.; Johnson, B.M.; Montgomery, S.A.; Peine, K.J.; Darr, D.B.; Yuan, H.; McKinnon, K.P.; et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 2018, 3, e120638. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef]
- Toraya-Brown, S.; Sheen, M.R.; Zhang, P.; Chen, L.; Baird, J.R.; Demidenko, E.; Turk, M.J.; Hoopes, P.J.; Conejo-Garcia, J.R.; Fiering, S. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine 2014, 10, 1273–1285. [Google Scholar] [CrossRef]
- Rancoule, C.; Magne, N.; Vallard, A.; Guy, J.B.; Rodriguez-Lafrasse, C.; Deutsch, E.; Chargari, C. Nanoparticles in radiation oncology: From bench-side to bedside. Cancer Lett. 2016, 375, 256–262. [Google Scholar] [CrossRef]
- Zhang, C.; Ni, D.; Liu, Y.; Yao, H.; Bu, W.; Shi, J. Magnesium silicide nanoparticles as a deoxygenation agent for cancer starvation therapy. Nat. Nanotechnol. 2017, 12, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Gao, J.; Pei, Q.; Xu, H.; Yu, H. Engineering Prodrug Nanomedicine for Cancer Immunotherapy. Adv. Sci. 2020, 7, 2002365. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xia, R.; Huang, Y.; Zhao, W.; Li, J.; Zhang, X.; Wang, P.; Venkataramanan, R.; Fan, J.; Xie, W.; et al. An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nat. Commun. 2016, 7, 13443. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Cheng, K.; Xu, Y.; Wang, Y.; Min, H.; Zhang, Y.; Zhao, X.; Zhao, R.; Anderson, G.J.; Ren, L.; et al. Modularly Designed Peptide Nanoprodrug Augments Antitumor Immunity of PD-L1 Checkpoint Blockade by Targeting Indoleamine 2,3-Dioxygenase. J. Am. Chem. Soc. 2020, 142, 2490–2496. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, N.; Zhang, L.; Calvo-Barreiro, L.; Kuncewicz, K.; Gabr, M. Inhibitors of Immune Checkpoints: Small Molecule- and Peptide-Based Approaches. J. Pers. Med. 2024, 14, 68. https://doi.org/10.3390/jpm14010068
Fuchs N, Zhang L, Calvo-Barreiro L, Kuncewicz K, Gabr M. Inhibitors of Immune Checkpoints: Small Molecule- and Peptide-Based Approaches. Journal of Personalized Medicine. 2024; 14(1):68. https://doi.org/10.3390/jpm14010068
Chicago/Turabian StyleFuchs, Natalie, Longfei Zhang, Laura Calvo-Barreiro, Katarzyna Kuncewicz, and Moustafa Gabr. 2024. "Inhibitors of Immune Checkpoints: Small Molecule- and Peptide-Based Approaches" Journal of Personalized Medicine 14, no. 1: 68. https://doi.org/10.3390/jpm14010068
APA StyleFuchs, N., Zhang, L., Calvo-Barreiro, L., Kuncewicz, K., & Gabr, M. (2024). Inhibitors of Immune Checkpoints: Small Molecule- and Peptide-Based Approaches. Journal of Personalized Medicine, 14(1), 68. https://doi.org/10.3390/jpm14010068