Abstract
Congenital heart diseases (CHDs) are structural or functional defects present at birth due to improper heart development. Current therapeutic approaches to treating severe CHDs are primarily palliative surgical interventions during the peri- or prenatal stages, when the heart has fully developed from faulty embryogenesis. However, earlier interventions during embryonic development have the potential for better outcomes, as demonstrated by fetal cardiac interventions performed in utero, which have shown improved neonatal and prenatal survival rates, as well as reduced lifelong morbidity. Extensive research on heart development has identified key steps, cellular players, and the intricate network of signaling pathways and transcription factors governing cardiogenesis. Additionally, some reports have indicated that certain adverse genetic and environmental conditions leading to heart malformations and embryonic death may be amendable through the activation of alternative mechanisms. This review first highlights key molecular and cellular processes involved in heart development. Subsequently, it explores the potential for future therapeutic strategies, targeting early embryonic stages, to prevent CHDs, through the delivery of biomolecules or exosomes to compensate for faulty cardiogenic mechanisms. Implementing such non-surgical interventions during early gestation may offer a prophylactic approach toward reducing the occurrence and severity of CHDs.
1. Introduction
The development of the human heart is a complex process that is initiated early during embryogenesis, orchestrated by intricate molecular and cellular events. Congenital heart diseases (CHDs), which result from improper embryonic development, are a group of structural and functional cardiac defects that affect approximately 1% of live births [1]. CHDs may present variable degrees of severity, ranging from minor defects with minimal or no clinical impact to severe malformations requiring immediate medical intervention at birth. Some cardiac conditions may necessitate lifelong treatment and care, frequently resulting in long-term morbidity and increased mortality rates [2]. At present, therapeutic approaches to the treatment of CHDs primarily focus on palliative interventions that improve cardiac structure and function, alleviate symptoms, and reduce the risk of long-term complications. Even surgical interventions, such as cardiac repair or transplantation, which significantly improve survival rates for patients, do not address the underlying genetic abnormalities associated with CHDs. As a result, the risk of CHDs in the children of patients who survived critical CHDs increases to 2–5% [1]. Consequently, there has been a shift from high infant mortality rates to increased survival of adults with complex CHDs, who are at risk for subsequent cardiovascular complications potentially leading to heart failure and the need for lifelong medical care [3]. In the United States, although pediatric admissions for patients with CHDs only account for 3.7% of total admissions, their annual cost is approximately $5.6 billion, which is about 15% of overall costs for pediatric patients [4]. Thus, CHDs are major and increasing health and economic burdens worldwide, and novel strategies to reliably diagnose, refine treatments for, and, if possible, prevent CHDs are urgently needed.
Current prenatal screening programs, genetic counselling, and optimization of maternal health and care, both before and during pregnancy, play a pivotal role in preventing CHDs [5]. The accuracy of prenatal tests to identify cardiac fetal anomalies and the development of innovative medical procedures have opened possibilities for fetal cardiac interventions (FCIs) in utero, at a critical developmental stage when the heart is still growing and developing. FCIs aim to correct fetal cardiac malformations that are either prone to progress with severe complications during mid- or late gestation, or to carry a high risk of fetal demise or life-threatening conditions at birth. FCIs also intend to improve neonatal and prenatal survival of the offspring, and limit lifelong morbidity and mortality [6,7,8,9]. Of interest, low maternal risks were reported with FCIs [10], but these interventions are currently limited to a small subset of CHDs (Table 1).
Thus, the limitations of surgical and medicinal treatments of cardiovascular diseases have stimulated the scientific and medical field to search for novel strategies. Consequently, human stem cells have emerged as a promising source of cells for cardiac regeneration in patients to complement current medical and surgical interventions, in addition to serving as cellular models to uncover the underlying mechanisms of CHDs [11,12]. To date, numerous preclinical and clinical studies have been performed to treat CHDs using embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and adult stem cells, such as cardiac progenitor cells and mesenchymal stem cells [13,14,15,16,17]. Although the outcomes of these trials remain largely unsatisfactory, the usage of autologous and allogenic stem cells to complement surgical interventions in infants with CHDs have recently shown positive results, particularly in patients with hypoplastic left heart syndrome (HLHS) [3,13,18,19,20]. To further reduce surgical interventions in patients, the tissue engineering research field is actively developing cell-seeded clinical patches that are able to either grow in synchronization with the cardiovascular structures or to be gradually replaced by the newly formed tissues of treated infants [3]. In addition to their differentiation abilities, stem cells secrete a wide range of bioactive molecules (secretomes) and release extracellular vesicles, including exosomes, which have promising potential in facilitating the repair and regeneration of damaged adult cardiac tissues and improving heart function [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. The secretomes and exosomes released by various cells have significant roles in (cardiac) development and tissue repair, and hold promising potential toward reversing CHDs, or at least mitigating their severity [19,37,38,39,40,41].
The causes of CHDs are often multifactorial and may involve inherited genetic mutations, sporadic developmental errors, and/or environmental factors [42,43,44,45,46]. Mutations or other genomic abnormalities have been shown to occur in genes that play crucial roles in cardiac development (Table 2), including genes encoding for transcriptional and epigenetic/chromatin remodeling factors (such as NKX2.5, TBX5, GATA4, CITED2, TBX20, p300, and CBP), cell signaling and adhesion proteins (such as ACVR1, NOTCH1, and PDGFRA), and structural sarcomere proteins (such as MYH6, MYH7, and ACTC1) [47,48]. Chromosomal anomalies resulting in syndromic complications, such as Down syndrome (trisomy 21), Turner syndrome (monosomy X), and DiGeorge syndrome (22q11.2 deletion), are often associated with increased risk of CHDs [47,48].

Table 2.
Genes frequently mutated and associated with congenital heart diseases.
Table 1.
Clinical trials related to congenital heart diseases and in utero applications.
Table 1.
Clinical trials related to congenital heart diseases and in utero applications.
| Clinical Trial Number | Type and Status | Condition | Intervention or Treatment | Population | Evaluation | Results |
|---|---|---|---|---|---|---|
| NCT03944837 | Interventional Single-group assignment; Status: recruiting. | Severe Fetal CHD | Transient maternal oxygen gas administration during echocardio-graphic and MRI imaging. | Pregnant woman ≥18 years old, with fetal diagnosis of specific CHDs and intention of prenatal treatment. | Brain growth and maturation to birth, improvement of postnatal neurodevelopmental issues, identification of CHD types likely to benefit from chronic maternal hyperoxygenation. | Not available |
| NCT01736956 | Interventional, prospective, non-randomi-zed clinical trial; Status: complete. | Aortic stenosis and evolving HLHS | Fetal aortic transuterine valvuloplasty, periventricular approach. Control group with standard prenatal and postnatal care. | Pregnant woman ≥ 16 years old, with a fetus with normal heart anatomy and severe aortic stenosis. | Safety and efficacy of in utero percutaneous balloon dilation of fetal aortic valve with severe stenosis determined by fetal mitral valve and left ventricle growth, survival, and neurodevelopmental status. | No results posted |
| NCT03147014 | Interventional; Status: complete. | Fetal HLHS, atrial septal aneurysm, aortic coarctation. | Cardiovascular response to maternal hyperoxygenation in fetal CHDs. All singleton fetuses with CHDs at all gestational ages are eligible. | Pregnant women with a fetus harboring CHDs diagnosed at various gestational weeks. | Participants received 10–15 min hyperoxygenation, assessed for middle cerebral artery pulsation; myocardial diastolic function; flow patterns across tricuspid, mitral valves, and ductus venosus; changes in heart output and ratio of right–left ventricle flow; flow at the aortic isthmus if aortic coarctation. | No results posted |
| EudraCT 2016-003181-12 * | Prospective cohort study; Status: complete. | Pregnant women with fetus at risk of pulmonary hypoplasia due to VSD/AVSD. | Sonographic assessment of pulmonary vascular reactivity following maternal hyperoxygenation. | Pregnant woman ≥18 years old, with a fetus at risk for neonatal persistent pulmonary hypertension; non-pregnant control. | Fetal echocardiographic doppler within the first 48 h of life to assess pulmonary vasculature prior to and after maternal hyperoxygenation to predict development of neonatal pulmonary hypertension. | [49] |
CHD—congenital heart disease; HLHS—hypoplastic left heart syndrome; VSD—ventricular septal defect; AVSD—atrioventricular septal defect. All data collected from clinicaltrials.gov, except (*) from clinicaltrialsregister.eu/, accessed on 13 August 2023.
Maternal/embryonic–fetal environmental factors, including maternal diabetes, medications taken during pregnancy, infections, alcohol or drug abuse, exposure to tobacco smoke, chemicals or toxins, may also increase the risk for CHDs [5,48]. Of interest, embryonic heart and placental development are interrelated and concomitant, sharing regulatory molecules and signaling pathways. Placental defects are also more common in pregnancies with CHDs, and the inadequate placental function can contribute to persistent cardiac defects postnatally [50,51,52].
Despite the complexity of embryonic heart formation and the high incidence of CHDs, cardiogenesis is a remarkably robust developmental process. Indeed, dysfunctions of more than one gene and/or environmental insults are often necessary to drastically impair heart development and lead to severe CHDs or death during gestation [53,54,55]. The resilience of this process certainly relies on the complex and evolutionary conserved gene regulatory network, which is orchestrated by key signaling pathways and transcription factors, that belong to families of proteins sometimes displaying overlapping or redundant functions [56]. Their robustness also depends on the existence of multiple sources of cardiac progenitors with functional redundancy, ensuring the production of heart cells even in the event of loss or defects in formation, expansion, and differentiation of certain progenitors [57,58,59]. For instance, despite ablation of over 50% of emerging First Heart Field (FHF) and Secondary Heart Field (SHF) cardiac progenitors at early stages of cardiogenesis, mouse embryos survived and developed into adult animals without any apparent cardiac abnormalities [58,60]. Therefore, the dysfunction or loss of cardiac progenitors and cardiomyocytes in mouse embryonic hearts can truly be compensated by alternative embryonic mechanisms, which include the expansion and migration of other unaffected cardiac cells [58,60,61,62,63].
Recent studies, using animal models and in vitro differentiation of human pluripotent stem cells, have also suggested that cardiac cell lineages are pre-determined during the early stages of development. Both positioning and functions of cardiac cells in the heart may be defined even before gastrulation, and orchestrated by multiple signaling pathways and environmental cues [57,64,65,66,67,68,69]. This idea is also supported by the ability of pluripotent stem cells to generate self-organizing cardiac organoids when provided with external cues from the extracellular matrix (ECM) and signaling molecules [70]. Moreover, in the absence of maternal tissues in vitro, human blastocysts attached to instructive supports and/or, stimulated by signaling molecules (such as WNT and NODAL/ACTIVIN), autonomously self-organize and originate structures resembling the proper embryo, with key landmarks of normal development, including bilaminar disc formation, primitive streak formation, lineage commitment, and extraembryonic annexes [71,72,73,74]. Therefore, future clinical strategies that would consistently provide correct developmental signals at early embryonic stages have the potential to compensate for defective mechanisms, thereby significantly reducing the incidence of CHDs.
Here, we review key molecular and cellular processes involved in early heart development. We also explore recent findings supporting the idea that the targeted delivery of biomolecules or exosomes during embryonic development, as early as the blastocyst stage, may compensate for faulty genes, signaling pathways, and cardiac progenitors, and potentially reduce the occurrence and/or severity of CHDs.
2. Navigating Early Mammalian Embryonic Heart and Placental Development
2.1. Placental Development
Placental and embryonic heart developments occur simultaneously, forming a placenta–heart axis, and sharing developmental pathways and common susceptibility to genetic defects [51]. Thus, the developing heart is highly vulnerable to early placental insufficiency. During early human embryogenesis, at embryonic days 4 to 5 (E4-5), the blastocyst attaches to the uterine wall. Trophoblast cells, encasing the inner cell mass that will originate the embryo proper, differentiate into inner cytotrophoblast cells, which are key for blastocyst implantation in the uterus, and stem cells that will also originate the outer syncytiotrophoblast [75,76]. The syncytiotrophoblast is responsible for the exchange of nutrients, gases, and waste products between the maternal and embryonic circulations, and also acts as a barrier against pathogens and harmful substances [77]. Interactions between the cytotrophoblast and syncytiotrophoblast are essential for proper placental development and function. Aberrant vascular placental development can lead to placental insufficiency and compromise fetal growth due to inefficient blood supply, and may result in preeclampsia, gestational diabetes, intrauterine growth restriction (IUGR), and placental abruption [78,79,80,81,82]. These complications are associated with an increased risk of adverse fetal outcomes, including CHDs [83,84,85,86]. Vascular endothelial growth factor (VEGF) and its receptors, hypoxia-inducible factors (HIFs), which regulate the cellular response to hypoxia, play a crucial role in placental and heart development, as well as in vascular remodeling, and their dysfunction increases the risk for CHDs [75,87,88,89]. Immune modulation is also essential for placental development and for fetal tolerance by the maternal immune system [90,91].
2.2. Heart Development
During embryogenesis, the heart is progressively built with multiple mature and functional cells, originating from many molecularly distinct progenitor cells that arise at different times and structures from the developing embryo [57,65,66,67,92,93]. The heart primarily develops from three main pools of embryonic progenitors, which are the FHF, SHF, and the proepicardium cells (Figure 1). The FHF and SHF derive from the earliest MESP1-marked cardiac mesodermal progenitors, which emerge shortly after gastrulation [57,64,65,66,67]. However, precardiac progenitor cells in the nascent mesoderm are, in fact, a heterogeneous population, composed of molecularly distinct progenitors that leave the primitive streak in a sequential manner, with contributions to specific and overlapping parts of the heart [93,94,95].
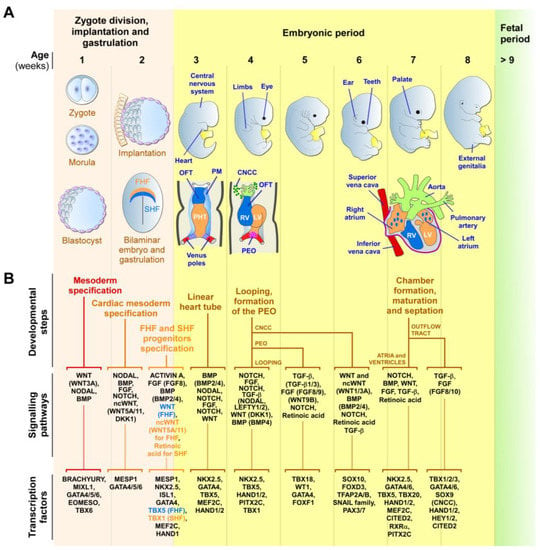
Figure 1.
Early human cardiac development. (A) Schematic representation of the zygotic and embryonic stages of human development, highlighting key developmental programs and milestones. Gestational times are indicated in weeks. The initial steps of heart, central nervous system, limb, eye, ear, tooth, and genital development are shown. The primary (FHF—blue) and secondary (SHF—orange) heart field cells, the cardiac neural crest cells (CNCCs—green), and the proepicardial organ (PEO—purple), and their respective derivatives, are illustrated. During week 2, the cardiac mesoderm gives rise to progenitors of the FHF and SHF, which form the cardiac crescent. The FHF originates the primary heart tube (PHT), which subsequently contributes to the left ventricle (LV) and parts of the right and left atria. SHF cells migrate through the pharyngeal mesoderm (PM) and contribute to the elongation of the PHT by ingression at both atrial and venous poles. SHF cells contribute to the development of the right ventricle (RV), outflow tract (OFT), atria, and inflow myocardium. Cells originating from the venous poles (red) give rise to the superior and inferior vena cava. The PEO cells contribute to the epicardium and coronary vessels. CNCCs migrate from the dorsal neural tube into the cardiac OFT, where they contribute to the formation of the septum, separating the truncus arteriosus into the aorta and pulmonary artery, as well as contributing to heart valve formation and parasympathetic innervation. (B) Overview of the secretomes, main signaling pathways, and transcription factors that regulate each listed stage of heart development. The noncanonical WNT pathway is indicated as ncWNT.
The first cells to leave the primitive streak, emerging at approximately E15-16 in humans, are FHF cells that give rise to the cardiac crescent structure, which migrates and fuses at the midline to form the primitive linear heart tube (at around E19 in humans) [42,57,64,65,66,93,96,97]. The linear heart tube, destined to generate the bulk of the atrial chambers and left ventricle, contains an outer myocardium and an inner endocardium, separated by a specific ECM known as the cardiac jelly. Rhythmic contractions are initiated by cardiomyocytes of the heart tube at around E21 in humans. At approximately E19-20, human SHF cells migrate into the heart tube and differentiate into cardiomyocytes, smooth muscle, and endothelial cells. The SHF contributes to the outflow tract (OFT), the right ventricular region, the atrioventricular canal structures, and the atria [42,57,64,65,66,93,96,97].
Around E28, the human heart tube undergoes a rightward looping to initiate the formation of the four-chambered heart. At this stage, the conductive system also begins to develop from cardiac progenitors, which differentiate into specialized myocytes with high conductance to form the sinoatrial node, atrioventricular node, bundle of His, and Purkinje fibers, rather than working cardiomyocytes [98,99]. As the heart develops, conduction cells establish connections to form a functional electrical system that coordinates the contraction and relaxation of the cardiac muscle. Further maturation and refinements of the conductive system occur throughout fetal development, with further structural and functional changes happening after birth and during postnatal growth [98,99]. In humans, ventricular septation starts around E50 and originates from the myocardium, which generates the ventricular septum and establishes the right and left atrio-ventricular canals. Atrial septation starts at E60 and derives from the septum primum and septum secundum, concomitantly with OFT septation [42,67,100,101].
OFT septation, which results in the formation of the aorta and the pulmonary artery, and their respective connections to the left and right ventricles, is accomplished by SHF cells and cardiac neural crest cells (CNCCs), a subpopulation of neural crest cells that delaminate from the neural tube [101,102]. CNCCs also give rise to the tip of the interventricular septum and OFT cushions, which will differentiate into the aortic and pulmonary valves and the parasympathetic coronary innervation [102]. The proepicardial organ is an extracardiac structure, which develops distinctly from the heart tube (at E9.5 in mouse) and contributes to the epicardial cells located around all heart chambers [95,103,104]. Other early multipotent progenitor cells, marked by KDR/FLK1/VEGFR2 expression, which are a source for endocardium, myocardium, and hematopoietic progenitors, were also detected in the primitive streak [57,105]. The major functional structures of the human heart are completed by E60. After this stage, the heart undergoes progressive growth, as well as structural, metabolic, and functional maturation processes, which are vital for its function during a lifetime [70].
3. Tracing the Roles of Signaling Pathways and Exosomes in Heart Development
3.1. Secretomes and Heart Development
Secretomes refer to a variety of secreted proteins, bioactive molecules, and factors released by cells that act locally to modulate cellular processes on neighboring cells, including processes necessary for development and tissue repair [30,106]. Among those factors, modulators of the WNT signaling pathway play a critical role in cardiac specification and differentiation, through stabilization and nuclear translocation of β-catenin, which, in turn, regulates the expression of cardiogenic genes [107,108,109,110,111,112,113]. NOTCH receptors and ligands are also important cardiogenic mediators through cell–cell communication, controlling the differentiation of cardiac progenitor cells into various cardiac lineages during heart development [114,115,116,117,118,119,120,121,122,123,124,125,126]. Bone morphogenetic proteins (BMPs), which are key regulators of embryonic development, promote the differentiation of cardiac mesoderm and regulate the formation of heart structures, through the expression of cardiac-specific transcription factors critical for cardiomyocyte differentiation [127,128,129,130,131,132,133,134,135,136,137]. The fibroblast growth factor (FGF) pathway is important for the formation of the OFT, ventricular maturation, and valve development, by modulating cell proliferation, survival, and differentiation [138,139,140]. Other secreted factors, such as insulin-like growth factor 1 (IGF-1) [141,142,143,144,145], VEGF [146,147,148,149,150], and transforming growth factor beta (TGF-β) family members, including ACTIVIN and NODAL [115,151,152,153,154], have been shown to enhance cardiomyocyte differentiation and maturation. Both ACTIVIN and NODAL crosstalk with other signaling molecules and modulate the activity of transcription factors to coordinate the specification, proliferation, and differentiation of cardiac progenitors during heart development (Figure 1B). Gastrulation is a critical embryonic step resulting in the formation of the three germ layers (endoderm, mesoderm, and ectoderm) and the specification of cell fates. During this process, mesodermal induction is stimulated by the convergent action of BMPs, NODAL/ACTIVIN, and canonical WNT signaling [93,155,156]. The emergence of mesodermal cells is marked by the expression of the transcription factor BRACHYURY and requires the activation of β-catenin by the canonical WNT pathway. Next, the inhibition of the WNT canonical pathway, achieved by proteins such as DKK1 and noncanonical WNT-related proteins, is critical for cardiac mesoderm specification [109,113,156,157,158]. Angiogenesis and vasculogenesis are also critical for cardiovascular development, as they ensure the formation of functional vascular networks through which to supply oxygen and nutrients to the developing heart. These processes are modulated by secreted proteins, such as VEGF, basic FGF, and angiopoietin-1, which are also capable of enhancing neovascularization in adult ischemic hearts [159,160,161,162].
3.2. Exosomes and Heart Development
Exosomes are formed through the inward budding of endosomal membranes and are subsequently released into the extracellular space to be captured by target cells [163]. The proteomic analysis of exosomes has revealed the presence of proteins involved in cell signaling, membrane transport, and ECM remodeling [164]. Additionally, exosomes may contain various types of messenger RNA (mRNA), microRNA (miRNA), long non-coding RNA (lncRNA), and mitochondrial miRNA [165,166,167], which can be transferred to recipient target cells and alter their gene expressions and cellular functions. During heart development, exosomes from multiple cell sources, including stem cells, have emerged as key mediators of intercellular communication in various biological contexts, including cardiac regeneration or repair (Table 3) [37,168,169,170]. For example, cardiac exosomes and secreted factors derived from cardiomyocytes, endothelial cells, or cardiac progenitor cells are important to the regulation of cardiac cell fate determination, proliferation, migration and differentiation, and heart morphogenesis [24,30,166,171,172,173]. Moreover, exosomes derived from cardiac and endothelial cells modulate epithelial-to-mesenchymal transition, favoring angiogenesis and the formation of coronary vessels, which are essential for heart development [174,175,176,177]. In addition, exosomes released through circulation during pregnancy via the umbilical cord, placenta, amniotic fluid, and amniotic membranes contribute to essential physiological functions in fetal–maternal communications, and to physiological processes such as angiogenesis, endothelial cell migration, and embryo implantation and placentation [178,179,180].
Table 3.
Examples of miRNA enriched in exosomes from various human cell sources, and their effect on the cardiovascular system.
Some exosomes, such as those derived either from diabetic or obese pregnant mice, cross the maternal–fetal barrier when injected through circulation and contribute to anomalies in cardiac development, such as ventricular septal defects, cardiac hypertrophy, pericardial effusions, and compromised systolic and diastolic functions [178,181,182,183]. Exosomes from the visceral adipose tissue of obese pregnant mice displayed an altered composition in miRNA, such as a decrease in miR-19b, which is involved in regulating inflammation and cardiac development, resulting in altered placental and cardiac functions [183]. Circulating exosomes, which are characteristic of maternal metabolic adaptations, have also been proposed to serve as pregnancy complication markers for gestational diabetes, hypertension, and pre-eclampsia, and to facilitate the determination of embryos with neural tube defects, CHDs, and other conditions, such as Down syndrome [178,184,185,186]. Overall, paracrine signaling mechanisms, mediated by exosomes and secreted factors, ensure the global regulation of cardiac morphogenesis [187], and their dysregulation is likely to result in CHDs [157,188,189,190,191].
4. Exploring the Impact of Diet and Medicinal Supplements on Heart Development
4.1. Diet Supplements and Heart Development
Both animal model studies and human trials have shown promising outcomes in using drugs and dietary supplementation as preventive measures for CHDs, setting the possibility to interfere with adverse cardiac defects through the intake or administration of biomolecules. Folic acid supplementation before and during pregnancy is a notable example, as it reduces the risk of neural tube defects and certain CHDs, particularly septal defects [192,193]. Supplementation of zinc (Zn) to diabetic mothers has also effectively reduced glucose-dependent oxidative stress, associated with cardiomyopathy in animals and humans, by preventing apoptosis in cardiomyocytes, and its supplementation at early stages of pregnancy was proposed to prevent CHDs induced by gestational diabetes [194]. Retinoic Acid (RA), a vitamin A derivative, is indispensable for many embryonic developmental processes, including heart formation (Figure 1), but the rigorous control of its production by cardiac embryonic tissues is necessary to prevent disastrous congenital malformations, such as anomalies in heart looping, the aortic arch, transposition of the great arteries, coronary defects, double-outlet right ventricle, myocardial hypoplasia, tetralogy of Fallot, and OFT and septal defects [195]. The injection of wild-type ESCs into blastocysts deficient in RA signaling successfully rescued various morphogenetic anomalies, such as left–right heart tube looping defects, through increased production of RA, which diffused to activate RA signaling in the defective cells [196]. This study demonstrated that endogenous embryonic production of RA can compensate for developmental heart defects. However, maternal supplementation was unable to provide the necessary levels of RA needed to rescue the mutants [196]. These examples indicate the possibility to limit CHDs by supplying biomolecules to mothers or embryos in pre- or early pregnancies. However, excessive fetal exposure to 13-cis-RA supplements (isotretinoin, Accutane) can induce CHDs. Alterations in fetal retinoid metabolism due to cross reactions with drugs (valproate), toxins (nitrofen, tobacco, alcohol), infections (rubella), or coexisting medical conditions (gestational diabetes) also favored CHDs [195]. Therefore, to prevent CHDs, it is crucial to have a thorough understanding of the roles and optimal levels of vitamin A, as well as of the factors that influence retinoid metabolism [197]. In general, a careful determination of the intake quantity of dietary supplements or drugs will be crucial to developing safe strategies for promoting normal heart development, achieving cardio-protection, and avoiding adverse effects, including CHDs themselves.
4.2. Cardiac Complications Due to Drugs Used to Treat Non-Cardiac Defects
Particular attention needs also to be given to supplements used for the treatment of non-cardiac problems, such as prenatal corticosteroid therapies used to stimulate fetal lung maturation in preterm births, which may affect fetal heart development and function in adulthood. Indeed, some studies in animal models have suggested that fetal exposure to glucocorticoids may cause changes in cardiomyocyte size and collagen deposition, and impair cardiac contractility, ultimately leading to an increase in the left ventricular mass and an alteration in cardiac function [198]. Maternal steroid therapy used to treat or prevent fetal heart block has also been shown to have undesirable side effects on the fetuses, such as alterations in brain development, oligohydramnios, growth restrictions, and constriction of the arterial duct, as well as diabetes mellitus, adrenal insufficiency, and psychosis for the mother [199,200]. Overall, several supplements administered before and during early pregnancy have shown potential for preventing CHDs and other congenital conditions. However, more research is required to determine the appropriate dosage of supplements in order to maximize the benefits for heart development while minimizing any adverse effect.
5. Unveiling the Potential of Secretomes and Exosomes for CHD Prevention
5.1. Exogenous Instructive Molecules to Mitigate CHDs
Multiple signaling pathways, which may display overlapping or redundant functions, are involved in generating cardiac progenitors and other cell lineages [57,64,65,66,67,68,69]. All issues considered, it is tempting to propose that the supplementation of exogenous cardiogenic instructive molecules at key moments during early embryonic stages may compensate for defective pathways and/or lineage decisions of cardiovascular progenitors to mitigate CHDs. To date, only a few experimental reports on animal models and stem cells support this idea. For instance, prolonged hypoxia causes DNA damage, premature senescence, impaired angiogenesis, and fibrosis associated with the upregulation of TGF-β1 expression in human fetuses with HLHS [201]. In vitro, hypoxia exposure of a human HLHS-derived iPSC disease cell model promotes the differentiation into cardiac fibroblasts instead of cardiac progenitors, cardiomyocytes, and endothelial cells [201]. Inhibition of TGF-β1 activity by its antagonist compound SB431542, in HLHS-derived iPSC exposed to hypoxia, prevented senescence and promoted genomic stability [201], suggesting that early interventions to inhibit TGF-β1 could improve ventricular growth and overcome pathways dysregulated in HLHS. IUGR is another gestational defect that hinders fetal growth in the uterus. IUGR is associated with increased placental vascular resistance, which forces the workload onto the fetal heart and increases the risk of cardiovascular disease [202]. Maternal administration of IGF-1 and IGF-2, which are known to stimulate placental and fetal growth in animal models, showed promising potential to treat IUGR in cases where downregulation of IGF-1 receptors in the placenta is not observed [202,203].
5.2. Exosomes, a Non-Cellular Approach for Correcting Embryonic Cardiac Defects
Emerging evidence suggests that the beneficial effects of stem cells in medical applications may primarily result from paracrine signaling molecules and the release of extracellular vesicles, rather than from the direct integration of stem cells or their derived cells into the target tissue [204]. Exosomes, which can be isolated and characterized by well-established approaches [205,206], present advantages such as scalable production, easy storage, consistent morphology and function, compliance with regulatory standards, and possible reduction in the variability of outcomes associated with cell therapies [207]. The therapeutic potential of exosomes and secretomes derived from hormonally primed human endometrial epithelial cells was tested in mouse models, and proven to enhance embryonic growth, development, and implantation [181]. This study also suggested that dysfunctions of the endometrial secretomes, which hinder implantation in cases of infertility, can be amended through the supply of exogenous exosomes. Also, maternal diabetic pregnancies in mouse models showed a higher occurrence of neural tube defects associated with exosomes produced from vascular progenitor cells expressing FLK1 derived from mesoderm cells lacking Survivin [208]. Interestingly, delivery into the amniotic cavity of Survivin-enriched vascular progenitor exosomes prevented neural tube defects in diabetic pregnancies [208], demonstrating the capacity of modified exosomes delivered in utero to limit neuronal pathology. Thus, exosomes are a promising non-cellular alternative to cells for clinical applications to correct embryonic defects. Exosomes can also be engineered to carry specific cargo, such as miRNAs or even gene-editing tools more suitable for the correction of genetic abnormalities associated with CHDs [176,209]. Notably, exosomes derived from the cardiac progenitors of children undergoing reconstructive heart surgeries showed promising effects in promoting angiogenesis, reducing fibrosis, resolving hypertrophy, and improving cardiac function in a rat model of heart arrhythmias caused by ischemia-reperfusion injury [39], showing the cardioprotective effect of exosomes from infant cardiac cells. Interestingly, exosomes from neonatal progenitors improved cardiac function regardless of oxygen levels, while exosomes from older children were only reparative in hypoxic conditions. Therefore, further research is needed to determine the most suitable cell sources and culture conditions for the production and modification of exosomes that would yield the best personalized clinical outcomes in the treatment of CHDs [40,208].
6. Harnessing Secreted Factors in Embryogenesis for Protection against CHDs
Like in many diseases, the early correction of a CHD is likely to result in a better clinical outcome, through early normalization of cardiogenesis. Reports on stem cell differentiation and heart development have suggested that cell positioning and functions within the heart may be determined prior to gastrulation, through precise signaling pathways and environmental cues [57,64,65,66,67,68,69]. Therefore, early interventions aiming to target the blastocyst at the pre-gastrulation/pre-implantation stage, very early embryonic states, may reduce the occurrence and severity of CHDs. Interestingly, pioneering studies on Inhibitors of Differentiation (ID) genes have revealed that double knockout of any pair of these genes in mouse models results in prominent cardiac defects and midgestational lethality of the embryos [210,211]. However, the injection of wild-type ESCs, either in ID-null blastocysts to form a chimera, or intraperitoneally in females prior to conception, rescued cardiac malformations and viability of ID-null embryos through upregulation of IGF-1 and WNT5A, without incorporation of wild-type ESCs into ID-null embryos [210,211]. Similarly, a chimeric embryo formation, using wild-type ESCs, improved the morphogenetic defects of RA signaling-deficient embryos by increasing RA expression [196]. Interestingly, transient exogenous WNT5A and WNT11, supplied at the 1-cell stage, efficiently rescued the viability and cardiac defects of CITED2-depleted zebrafish embryos and cardiac differentiation of mouse ESCs [212]. Dysfunctions of CITED2, a transcriptional modulator, have been widely associated with zebrafish, mouse, and human CHDs, as well as with embryonic lethality [55,212,213,214,215,216,217,218,219,220]. At the cellular level, CITED2 regulates the expressions of numerous genes involved in early cardiogenesis, such as BRACHYURY, MESP1, ISL1, GATA4, TBX5, MEF2C, NODAL, LEFTY1/2, PITX2C, VEGFA, WNT5A, and WNT11, among others [212,213,214]. Thus, the supplementation of WNT5A and WNT11 at the blastocyst stage in utero holds potential for rescuing CHDs associated with CITED2 dysfunction in mammals, since the function of CITED2 is conserved across vertebrates [212,214,216,221]. This strategy may also prevent CHDs triggered by the dysfunction of many genes other than CITED2, such as CITED2-target cardiogenic genes, including WNT5A and WNT11.
Moreover, WNT5A and WNT11 trigger many congenital cardiac anomalies, and contribute to DiGeorge syndrome, when defective [222,223,224,225,226,227,228]. Indeed, the WNT5A and WNT11 proteins are central for proper gestation and successful pregnancy, and their expressions are naturally highly increased and localized in the uterine luminal epithelium prior to and during blastocyst attachment to the uterus [229,230]. These proteins also promote embryonic uterine implantation and survival [229], as well as placental growth [230]. WNT5A and WNT11 are also important in late gastrulation, for the regulation of the anterior–posterior axis elongation, notochord extension, and proper patterning of the neural tube and somites [231]. In early mouse gastrulation, WNT11 is initially expressed in endoderm progenitors, and later, during mid-gastrulation, it plays a role in the formation of the embryonic and extraembryonic endothelia, as well as the formation of the endocardium in all chambers of the developing heart [232]. During late gastrulation, WNT11 showed successive waves of expression in different regions of the myocardium, important to originate left ventricle precursors (FHF progenitors) from E7.0–8.0, right ventricle progenitors (SHF progenitors) from E8.0–9.0, and the superior wall of the OFT from E8.5–10.5 (also SHF progenitors) [232]. Other studies have implicated WNT5A and WNT11 in the development of cardiac progenitors in vitro and in vivo [233,234,235,236], proper fetal hematopoiesis [237,238], kidney development [239], and guidance of sympathetic neurons to their innervation targets in vivo [240], among other processes. Altogether, the broad range of effects exhibited by WNT5A and WNT11 during early development emphasizes their high potential in limiting CHDs and other birth abnormalities when supplied exogenously at early embryonic stages. Another recent study highlighted the role of a subset of human amniotic epithelial cells (AECs) in mesoderm formation at E8.0–9.0 during early post-implantation stages [241]. Interestingly, impaired mesoderm formation and lethality due to loss of ISL1 expression in non-human primate embryos, or in human AEC differentiation in vitro due to the decrease in BMP4 expression, was partially restored through supplementation of BMP4 [241]. Overall, these findings demonstrate the potential for biomolecules, such as WNT5A/WNT11, BMP4, and inhibitors of the TGF-β pathway (SB431542, for instance), to reduce CHDs when administered at very early stages of development.
7. Challenges and Ethical Considerations
The experiments on mouse ID-null and RA-defective embryos rescued by the introduction of ESCs, either in defective blastocysts or in the mother prior to gestation [196,210,211], are groundbreaking studies, establishing a proof-of-principle that early interventions have the potential to restore normal development to genetically defective blastocysts. However, the translation of such strategies to humans, based on pluripotent stem cell delivery into blastocysts, remains both ethically and technically undesirable. Indeed, the alteration of such early human embryos, even for health purposes, is per se questionable. Moreover, the transplantation of human pluripotent stem cells (ESCs or iPSCs) in a clinical setting carries high risks of tumor formation and differentiation into unwanted cells/tissues [242]. Nevertheless, pre-implanted blastocysts are promising targets for medical interventions, since their size and localization, near or in the uterine lumen, may facilitate the delivery and effective diffusion of corrective biomolecules. Moreover, blastocysts exhibit minimal tissue commitment bias, making them possibly more responsive to exogenous cues aiming to correct congenital defects. In both studies presented above, the corrective effects of wild-type ESCs were contributed by the neomorphic effects of factors secreted by these cells, such as WNT5A, IGF-1, and RA [196,210,211]. These findings strongly support the idea that exogenous administration of WNT5A and WNT11, as well as IGF-1 and possibly other factors to be identified, to early embryos has the capacity to substitute for pluripotent stem cell transplantation.
The intricate expression patterns and multiple functions of the WNT5A, WNT11, IGF-1, BMP4, and TGF-β pathways during placental and embryonic development portend that any disturbance in their expression and signaling network may have drastic impacts on normal embryonic development. However, the supplementation of these factors may only need to be temporary and administered at very specific times, as remarkably suggested by the transient supply of exogenous WNT5A/WNT11 recombinant proteins at the onset of mouse ESC differentiation for 2 days, or at the 1-cell stage in zebrafish embryos to rescue the cardiogenic defects and lethality caused by CITED2 depletion [212]. Moreover, compelling evidence suggests that the exogenous administration of WNT5A and WNT11 proteins to early embryos may not only be well-tolerated, but may even provide general beneficial effects on development, rather than potential adverse effects. Indeed, WNT5A is naturally and abundantly expressed in the endometrium of mammals during the pre-implantation period, ensuring proper placental and embryonic development from the morula to the blastocyst stage, safeguarding the success of pregnancies [229,230,243,244,245,246,247]. Therefore, the supply of exogenous WNT5A to the uterine lumen before implantation coincides with its naturally high expression during this period, suggesting that the organism may be prepared to handle abundant levels of WNT5A and may mitigate any potential adverse effects caused by its exogenous administration. In addition, WNT5A and WNT11 form protein complexes, which were proposed to be more effective in activating their cellular functions than the individual proteins were [248]. Thus, the combined administration of WNT5A/WNT11 may be more inclined to fulfil the rescuing effects and buffer the adverse effects. Accordingly, the combined supplementation of WNT5A/WNT11 was tendentially more efficient for rescuing the viability and cardiac anomalies of CITED2-deficient zebrafish embryos, and for restoring the expression levels of early cardiogenic factors (such as BRACHYURY, MESP1, and ISL1) to control levels in CITED2-depleted mouse ESCs [212]. Also of interest, the combined administration of WNT5A/WNT11 to wild-type zebrafish embryos additionally reduced natural occurrences of death, cardiac anomalies, and variability in heart rate, rather than showing adverse effects [212]. Together, these observations suggest that the temporary supply of WNT5A/WNT11 in the uterine lumen during peri-implantation, to target the blastocyst, would meet tolerant conditions, bearing increased WNT5A/WNT11 levels able to prevent CHDs with no or minimal side effects. Further investigation is needed to determine whether other cardiogenic molecules, such as BMP4 or antagonists of TGF-β, IGF-1, VEGF, and HGF, for example [106,160,249,250,251], as well as relevant exosomes, would also be able to limit CHDs, if delivered at the pre-implantation period.
Given the multifaceted nature of CHDs, and the intricate cellular interactions involved in cardiogenesis, there is potential for synergistic and amplified therapeutic effects by combining approaches, such as exosomes/secretomes and drug delivery, with gene therapy and/or tissue engineering. Furthermore, personalized medicinal strategies that consider individual genetic and molecular profiles would help to tailor treatments and improve care for patients with specific genetic factors associated with CHDs [28,252,253]. Future challenges also include standardization and optimization of secreted protein/exosome production, and full comprehension of their complex interplay with recipient cells to translate preclinical findings into safe clinical applications [254,255,256].
Finally, the reconsideration of ethical guidelines by the pillars of ethical governance, to shape responsibility and trust, is necessary to avoid the offer of defiant, unsafe, and unproven treatments by unauthorized clinics, similar to the existing issue in the field of stem cell-based therapies [257,258,259]. Indeed, the excitement and expectations surrounding such novel medical approaches may lead individuals at high risk of having children with CHDs to seek unapproved therapies, invoking the “right to try” concept. Therefore, balancing individual autonomy with regulatory oversight is essential to protecting patients’ well-being. Additionally, equitable access to these potential treatments should be ensured, regardless of socio-economic status, geographic location, or other discriminative barriers. Lastly, a responsible approach to research, including clinical trials, is necessary for the future use of these approaches when no alternative options are available, aligning with the ethical principle of proportionality.
8. Conclusions and Perspectives
Mammalian heart development is a complex process, involving distinct pools of cardiac progenitor cells located in specific regions of the embryo. The precise orchestration of the progenitor cells’ emergence in embryonic time and space, as well as their proliferation, migration, and differentiation process, which is stimulated by environmental signals and pathways, is crucial for proper embryonic heart assembly. Despite high complexity, the cardiac developmental process remains robust, probably due to overlap and redundancy of the main signaling pathways, and of the multiple cardiac progenitors driving cardiogenesis. However, the dysfunction of cardiac progenitor cells, and/or the disruption of molecular mechanisms underlying cardiogenesis, results in cardiac malformations in 1 out of 100 human births, and often in embryonic death during gestation. Despite prenatal screening programs and better genetic counselling, which play a crucial role in preventing these malformations, CHDs remain the most prevalent congenital anomalies worldwide.
The current therapeutic approaches for severe CHDs, which are primarily surgical, remain unsatisfying despite tremendous medical and technological progress. Indeed, interventions are mostly palliative and often result in a lifelong health burden for patients. Moreover, patients are typically treated for the first time during the peri- or prenatal period, when the heart has fully developed from deficient early embryonic processes. Thus, earlier interventions in embryonic development could mitigate the impact of faulty heart development and reduce the occurrence of congenital abnormalities. Only recently, FCIs have been implemented for a reduced subset of CHD cases linked to hypoplastic left heart, to correct cardiac malformations in utero during pregnancy. FCIs have yielded promising results, and, compared to conventional surgical interventions after birth, FCIs offer the advantage of intervening at a critical developmental stage, when the heart is still growing and developing. These early corrections aim to address the underlying issues and abnormalities in cardiac structure and function before they become more severe, or irreversible.
Decades of cardiovascular research, encompassing studies on animal and cell models, as well as human genetic investigations, have uncovered the fundamental mechanisms that drive heart development and CHDs. Importantly, these findings have revealed the existence of multiple cardiac progenitors, as well as key transcription factors and signaling pathways involved in proper heart development. The cardiogenic process is characterized by overlapping and redundant functions existing among cardiac progenitors, signaling pathways, and factors. Redundant features in cardiac development enable the heart to sometimes compensate for defects in progenitors or genetic abnormalities through alternative mechanisms. Thus, despite the imperfect nature of the cardiogenic system, most viable individuals have a functional heart with minimal defects that can sustain them from gestational life through adulthood. Most importantly, these findings highlight the potential to artificially activate embryonic genetic and environmental cues to overcome defective cardiogenic events and compensate for adverse heart developmental conditions in utero.
Recent reports have emphasized the potential of specific biomolecules, including WNT5A, WNT11, IGF-1, BMP4, and exosomes, to interfere with abnormal cardiac development and correct cardiogenesis, when administered exogenously at early stages of development. Among those, WNT5A and WNT11 are particularly interesting, since they have shown corrective properties in mouse and fish models. WNT5A and WNT11 are necessary for correct development of the heart and placenta, among other organs and tissues, and they are abundantly expressed in the uterine epithelium at peri-implantation, a key period when blastocysts reach the uterine lumen. In addition, the blastocyst may be the target of choice for WNT5A and WNT11 corrective effects, since these early embryonic structures may be responsive to the exogenous instructions necessary to originate functional pre-gastrulation cells/progenitors, defined to derive specific parts of heart structures. Thus, it is of interest to fully explore the potential of WNT5A and WNT11 for the development and implementation of future preventive therapies for CHDs.
Nevertheless, further understanding of developmental mechanisms, as well as dosages, functions, and cross-reactions of therapeutic molecules, is key to devise the best and safest approaches to use all biomolecules and proteins (including WNT5A, WNT11, IGF-1, and BMP4), as well as exosomes, for CHD prevention. Additional research on genetic and environmental cues, as well as the mechanisms overcoming defective cardiogenic events, may help to identify other molecules and exosomes and develop safe strategies through which to compensate for adverse heart developmental conditions. Implementing such non-surgical interventions during early gestation offers the potential for reducing the occurrence and severity of CHDs, as well as an early preventive measure in utero. Although the underlying genetic mechanism potentially leading to CHDs would not be addressed with those approaches, it would be of interest to explore such combinatorial strategies, since they may provide prophylactic options to prevent or limit the severity of CHDs.
Author Contributions
Conceptualization, J.B.; writing—original draft preparation, J.B.; writing—review and editing, J.B., R.P., B.S., H.S.L., M.T.F. and N.M.; project administration, J.B.; funding acquisition, J.B. and M.T.F. All authors have read and agreed to the published version of the manuscript.
Funding
J.B. was supported by Fundação para a Ciência e Tecnologia (FCT) and the Comissão de Coordenação e Desenvolvimento Regional do Algarve (CCDR Algarve), for financial support to the project ALG-45-2020-41; M.T.F. was also supported by FCT for the project 2022.09209.PTDC; B.S. received a FCT PhD fellowship, with the reference 2022.13253.BDANA; J.B., R.P., H.S.L., and N.M. were supported by the Algarve Biomedical Center (ABC) award—“Bolsa de investigação translacional—José Mariano Gago by ABC”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This article is dedicated to the memory of Laura A. Bragança (12/05/2003–10/05/2019), who was always a strong supporter of our work, and to all children suffering from congenital heart disease. We thank the Fundação para a Ciência e Tecnologia (FCT) and the Comissão de Coordenação e Desenvolvimento Regional do Algarve (CCDR Algarve) for financial support to the projects ALG-45-2020-41 to J.B. and FCT 2022.09209.PTDC to M.T.F., and the PhD fellowships 2022.13253.BDANA to B.M.S. and 2023.00861.BD to R.P. We also thank the Algarve Biomedical Center (ABC) for the award—“Bolsa de investigação translacional—José Mariano Gago by ABC” to J.B.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, S.M.; Devine, O.J.; Kucik, J.E.; Oster, M.E.; Riehle-Colarusso, T.; Nembhard, W.N.; Xu, P.; Correa, A.; Jenkins, K.; Marelli, A.J. Congenital Heart Defects in the United States. Circulation 2016, 134, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Rajamarthandan, S.; Francis, B.; O’Leary-Kelly, M.K.; Sinha, P. Update on stem cell technologies in congenital heart disease. J. Card. Surg. 2020, 35, 174–179. [Google Scholar] [CrossRef]
- Chowdhury, D.; Johnson, J.N.; Baker-Smith, C.M.; Jaquiss, R.D.B.; Mahendran, A.K.; Curren, V.; Bhat, A.; Patel, A.; Marshall, A.C.; Fuller, S.; et al. Health Care Policy and Congenital Heart Disease: 2020 Focus on Our 2030 Future. J. Am. Heart Assoc. 2021, 10, e020605. [Google Scholar] [CrossRef]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. Noninherited Risk Factors and Congenital Cardiovascular Defects: Current Knowledge. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef]
- Freud, L.R.; Tworetzky, W. Fetal interventions for congenital heart disease. Curr. Opin. Pediatr. 2016, 28, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Halder, V.; Ghosh, S.; Shrimanth, Y.S.; Manoj, R.K.; Mandal, B.; Thingnam, S.K.S.; Kumar, R. Fetal cardiac intervention and fetal cardiac surgery: Where are we in 21st century? Am. J. Cardiovasc. Dis. 2021, 11, 642–646. [Google Scholar]
- Friedman, K.G.; Tworetzky, W. Fetal cardiac interventions: Where do we stand? Arch. Cardiovasc. Dis. 2020, 113, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Gellis, L.; Tworetzky, W. The boundaries of fetal cardiac intervention: Expand or tighten? Semin. Fetal Neonatal Med. 2017, 22, 399–403. [Google Scholar] [CrossRef]
- Moon-Grady, A.J.; Morris, S.A.; Belfort, M.; Chmait, R.; Dangel, J.; Devlieger, R.; Emery, S.; Frommelt, M.; Galindo, A.; Gelehrter, S.; et al. International Fetal Cardiac Intervention Registry: A Worldwide Collaborative Description and Preliminary Outcomes. J. Am. Coll. Cardiol. 2015, 66, 388–399. [Google Scholar] [CrossRef]
- Huang, J.; Feng, Q.; Wang, L.; Zhou, B. Human Pluripotent Stem Cell-Derived Cardiac Cells: Application in Disease Modeling, Cell Therapy, and Drug Discovery. Front. Cell Dev. Biol. 2021, 9, 655161. [Google Scholar] [CrossRef]
- Terashvili, M.; Bosnjak, Z.J. Stem Cell Therapies in Cardiovascular Disease. J. Cardiothorac. Vasc. Anesth. 2019, 33, 209–222. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Oikonomou, E.K.; Moris, D.; Schizas, D.; Economopoulos, K.P.; Mylonas, K.S. Stem Cell Therapy for Congenital Heart Disease. Circulation 2017, 136, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Oh, H. Cell Therapy Trials in Congenital Heart Disease. Circ. Res. 2017, 120, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.-W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef]
- Bragança, J.; Lopes, J.A.; Mendes-Silva, L.; Santos, J.M.A. Induced pluripotent stem cells, a giant leap for mankind therapeutic applications. World J. Stem Cells 2019, 11, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.D.; Ferreira, A.; Fernandes, M.T.; Silva, B.M.; Esteves, F.; Leitão, H.S.; Bragança, J.; Calado, S.M. Human Stem Cells for Cardiac Disease Modeling and Pre-clinical and Clinical Applications—Are We on the Road to Success? Cells 2023, 12, 1727. [Google Scholar] [CrossRef] [PubMed]
- Trac, D.; Maxwell, J.T.; Brown, M.E.; Xu, C.; Davis, M.E. Aggregation of Child Cardiac Progenitor Cells Into Spheres Activates Notch Signaling and Improves Treatment of Right Ventricular Heart Failure. Circ. Res. 2019, 124, 526–538. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, S.; Shen, W.-B.; Kaushal, S.; Yang, P. The current status and future of cardiac stem/progenitor cell therapy for congenital heart defects from diabetic pregnancy. Pediatr. Res. 2018, 83, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Chase, D.M.; Gallicchio, V.S. The effect of stem cells in congenital heart disease. Clin. Med. Investig. 2019, 4, 1–12. [Google Scholar] [CrossRef]
- Femminò, S.; Bonelli, F.; Brizzi, M.F. Extracellular vesicles in cardiac repair and regeneration: Beyond stem-cell-based approaches. Front. Cell Dev. Biol. 2022, 10, 996887. [Google Scholar] [CrossRef] [PubMed]
- de Abreu, R.C.; Fernandes, H.; da Costa Martins, P.A.; Sahoo, S.; Emanueli, C.; Ferreira, L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat. Rev. Cardiol. 2020, 17, 685–697. [Google Scholar] [CrossRef]
- Ibrahim, A.; Marbán, E. Exosomes: Fundamental Biology and Roles in Cardiovascular Physiology. Annu. Rev. Physiol. 2016, 78, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Losordo, D.W. Exosomes and Cardiac Repair after Myocardial Infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef]
- Andriolo, G.; Provasi, E.; Lo Cicero, V.; Brambilla, A.; Soncin, S.; Torre, T.; Milano, G.; Biemmi, V.; Vassalli, G.; Turchetto, L.; et al. Exosomes from Human Cardiac Progenitor Cells for Therapeutic Applications: Development of a GMP-Grade Manufacturing Method. Front. Physiol. 2018, 9, 1169. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Ma, X.; Lin, S.; Wu, B.; Chen, Y.; Peng, C. Bone marrow mesenchymal stem cell-derived exosomes protect against myocardial infarction by promoting autophagy. Exp. Ther. Med. 2019, 18, 2574–2582. [Google Scholar] [CrossRef]
- Cai, C.; Guo, Y.; Teng, L.; Nong, Y.; Tan, M.; Book, M.J.; Zhu, X.; Wang, X.L.; Du, J.; Wu, W.J.; et al. Preconditioning Human Cardiac Stem Cells with an HO-1 Inducer Exerts Beneficial Effects After Cell Transplantation in the Infarcted Murine Heart. Stem Cells 2015, 33, 3596–3607. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Li, T.; Gu, J.; Yang, O.; Wang, J.; Wang, Y.; Kong, J. Bone Marrow Mesenchymal Stem Cell-Derived Exosomal miRNA-29c Decreases Cardiac Ischemia/Reperfusion Injury Through Inhibition of Excessive Autophagy via the PTEN/Akt/mTOR Signaling Pathway. Circ. J. Off. J. Jpn. Circ. Soc. 2020, 84, 1304–1311. [Google Scholar] [CrossRef]
- Ibrahim, A.G.-E.; Cheng, K.; Marbán, E. Exosomes as Critical Agents of Cardiac Regeneration Triggered by Cell Therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef]
- Maleki, B.; Alani, B.; Tamehri Zadeh, S.S.; Saadat, S.; Rajabi, A.; Ayoubzadeh, S.M.J.; Verdi, J.; Farrokhian, A.; Ghanbarian, H.; Noureddini, M.; et al. MicroRNAs and exosomes: Cardiac stem cells in heart diseases. Pathol. Res. Pract. 2022, 229, 153701. [Google Scholar] [CrossRef]
- Xia, W.; Chen, H.; Xie, C.; Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 2020, 12, 8241–8260. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, X.; Zhang, X.; Sun, Y.; Yan, Y.; Shi, H.; Zhu, Y.; Wu, L.; Pan, Z.; Zhu, W.; et al. Human umbilical cord mesenchymal stem cell exosomes enhance angiogenesis through the Wnt4/β-catenin pathway. Stem Cells Transl. Med. 2015, 4, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, X.; Cao, W.; Ma, J.; Sun, L.; Qian, H.; Zhu, W.; Xu, W. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Relieve Acute Myocardial Ischemic Injury. Stem Cells Int. 2015, 2015, 761643. [Google Scholar] [CrossRef] [PubMed]
- Zarà, M.; Amadio, P.; Campodonico, J.; Sandrini, L.; Barbieri, S.S. Exosomes in Cardiovascular Diseases. Diagnostics 2020, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Tseliou, E.; Fouad, J.; Reich, H.; Slipczuk, L.; de Couto, G.; Aminzadeh, M.; Middleton, R.; Valle, J.; Weixin, L.; Marbán, E. Fibroblasts Rendered Antifibrotic, Antiapoptotic, and Angiogenic by Priming with Cardiosphere-Derived Extracellular Membrane Vesicles. J. Am. Coll. Cardiol. 2015, 66, 599–611. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.S.; Benedict, C.; et al. Embryonic Stem Cell–Derived Exosomes Promote Endogenous Repair Mechanisms and Enhance Cardiac Function Following Myocardial Infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Chen, K.; Liang, J.; Qin, T.; Zhang, Y.; Chen, X.; Wang, Z. The Role of Extracellular Vesicles in Embryo Implantation. Front. Endocrinol. 2022, 13, 809596. [Google Scholar] [CrossRef]
- Agarwal, U.; George, A.; Bhutani, S.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Mehta, Y.; Platt, M.O.; Liang, Y.; Sahoo, S.; et al. Experimental, Systems, and Computational Approaches to Understanding the MicroRNA-Mediated Reparative Potential of Cardiac Progenitor Cell–Derived Exosomes From Pediatric Patients. Circ. Res. 2017, 120, 701–712. [Google Scholar] [CrossRef]
- Hoffman, J.R.; Park, H.-J.; Bheri, S.; Jayaraman, A.R.; Davis, M.E. Comparative computational RNA analysis of cardiac-derived progenitor cells and their extracellular vesicles. Genomics 2022, 114, 110349. [Google Scholar] [CrossRef]
- Yin, X.; Jiang, L.-H. Extracellular vesicles: Targeting the heart. Front. Cardiovasc. Med. 2023, 9, 1041481. [Google Scholar] [CrossRef]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.B.; Foo, S.Y.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Spero, T.L.; Nolte, C.G.; Garcia, V.C.; Lin, Z.; Romitti, P.A.; Shaw, G.M.; Sheridan, S.C.; Feldkamp, M.L.; Woomert, A.; et al. Projected Changes in Maternal Heat Exposure During Early Pregnancy and the Associated Congenital Heart Defect Burden in the United States. J. Am. Heart Assoc. 2019, 8, e010995. [Google Scholar] [CrossRef]
- Morton, S.U.; Quiat, D.; Seidman, J.G.; Seidman, C.E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 2021, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Pierpont Mary, E.; Brueckner, M.; Chung Wendy, K.; Garg, V.; Lacro Ronald, V.; McGuire Amy, L.; Mital, S.; Priest James, R.; Pu William, T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Blue, G.M.; Kirk, E.P.; Sholler, G.F.; Harvey, R.P.; Winlaw, D.S. Congenital heart disease: Current knowledge about causes and inheritance. Med. J. Aust. 2012, 197, 155–159. [Google Scholar] [CrossRef]
- McHugh, A.; El-Khuffash, A.; Bussmann, N.; Doherty, A.; Franklin, O.; Breathnach, F. Hyperoxygenation in pregnancy exerts a more profound effect on cardiovascular hemodynamics than is observed in the nonpregnant state. Am. J. Obstet. Gynecol. 2019, 220, 397.e1–397.e8. [Google Scholar] [CrossRef]
- Courtney, J.A.; Cnota, J.F.; Jones, H.N. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Maslen, C.L. Recent Advances in Placenta–Heart Interactions. Front. Physiol. 2018, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Rychik, J.; Savla, J.J. The placenta as the window to congenital heart disease. Curr. Opin. Cardiol. 2021, 36, 56–60. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Jongbloed, M.R.M.; Poelmann, R.E.; Bartelings, M.M. Structural Heart Disease: Embryology. In Fetal Therapy: Scientific Basis and Critical Appraisal of Clinical Benefits, 2nd ed.; Johnson, A., Oepkes, D., Kilby, M.D., Eds.; Cambridge University Press: Cambridge, UK, 2020; pp. 110–122. [Google Scholar] [CrossRef]
- Moreau, J.L.M.; Kesteven, S.; Martin, E.M.M.A.; Lau, K.S.; Yam, M.X.; O’Reilly, V.C.; del Monte-Nieto, G.; Baldini, A.; Feneley, M.P.; Moon, A.M.; et al. Gene-environment interaction impacts on heart development and embryo survival. Development 2019, 146, dev172957. [Google Scholar] [CrossRef]
- Bentham, J.; Michell, A.C.; Lockstone, H.; Andrew, D.; Schneider, J.E.; Brown, N.A.; Bhattacharya, S. Maternal high-fat diet interacts with embryonic Cited2 genotype to reduce Pitx2c expression and enhance penetrance of left-right patterning defects. Hum. Mol. Genet. 2010, 9, 3394–3401. [Google Scholar] [CrossRef]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Sturzu, A.C.; Rajarajan, K.; Passer, D.; Plonowska, K.; Riley, A.; Tan, T.C.; Sharma, A.; Xu, A.F.; Engels, M.C.; Feistritzer, R.; et al. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation 2015, 132, 109–121. [Google Scholar] [CrossRef]
- Sharma, B.; Ho, L.; Ford, G.H.; Chen, H.I.; Goldstone, A.B.; Woo, Y.J.; Quertermous, T.; Reversade, B.; Red-Horse, K. Alternative Progenitor Cells Compensate to Rebuild the Coronary Vasculature in Elabela- and Apj-Deficient Hearts. Dev. Cell 2017, 42, 655–666. [Google Scholar] [CrossRef]
- Drenckhahn, J.-D.; Schwarz, Q.P.; Gray, S.; Laskowski, A.; Kiriazis, H.; Ming, Z.; Harvey, R.P.; Du, X.-J.; Thorburn, D.R.; Cox, T.C. Compensatory Growth of Healthy Cardiac Cells in the Presence of Diseased Cells Restores Tissue Homeostasis during Heart Development. Dev. Cell 2008, 15, 521–533. [Google Scholar] [CrossRef]
- Founta, K.-M.; Papanayotou, C. In Vivo Generation of Organs by Blastocyst Complementation: Advances and Challenges. Int. J. Stem Cells 2022, 15, 113–121. [Google Scholar] [CrossRef]
- Coppiello, G.; Barlabé, P.; Moya-Jódar, M.; Abizanda, G.; Barreda, C.; Iglesias, E.; Linares, J.; Arellano-Viera, E.; Ruiz-Villalba, A.; Larequi, E.; et al. In vivo generation of heart and vascular system by blastocyst complementation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kitajima, S.; Takagi, A.; Inoue, T.; Saga, Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development 2000, 127, 3215–3226. [Google Scholar] [CrossRef] [PubMed]
- Houyel, L.; Meilhac, S.M. Heart Development and Congenital Structural Heart Defects. Annu. Rev. Genom. Hum. Genet. 2021, 22, 257–284. [Google Scholar] [CrossRef]
- Tyser, R.C.V.; Ibarra-Soria, X.; McDole, K.; Arcot Jayaram, S.; Godwin, J.; van den Brand, T.A.H.; Miranda, A.M.A.; Scialdone, A.; Keller, P.J.; Marioni, J.C.; et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 2021, 371, eabb2986. [Google Scholar] [CrossRef] [PubMed]
- Swedlund, B.; Lescroart, F. Cardiopharyngeal Progenitor Specification: Multiple Roads to the Heart and Head Muscles. Cold Spring Harb. Perspect. Biol. 2020, 12, a036731. [Google Scholar] [CrossRef]
- Kloesel, B.; DiNardo, J.A.; Body, S.C. Cardiac Embryology and Molecular Mechanisms of Congenital Heart Disease—A Primer for Anesthesiologists. Anesth. Analg. 2016, 123, 551–569. [Google Scholar] [CrossRef]
- Lawson, K.A.; Meneses, J.J.; Pedersen, R.A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 1991, 113, 891–911. [Google Scholar] [CrossRef]
- Scialdone, A.; Tanaka, Y.; Jawaid, W.; Moignard, V.; Wilson, N.K.; Macaulay, I.C.; Marioni, J.C.; GÃttgens, B. Resolving early mesoderm diversification through single-cell expression profiling. Nature 2016, 535, 289. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, P.; Jahnel, S.M.; Mendjan, S. In vitro models of the human heart. Development 2021, 148, dev199672. [Google Scholar] [CrossRef]
- Martyn, I.; Kanno, T.Y.; Ruzo, A.; Siggia, E.D.; Brivanlou, A.H. Self-organization of a human organizer by combined Wnt and Nodal signalling. Nature 2018, 558, 132–135. [Google Scholar] [CrossRef]
- Deglincerti, A.; Croft, G.F.; Pietila, L.N.; Zernicka-Goetz, M.; Siggia, E.D.; Brivanlou, A.H. Self-organization of the in vitro attached human embryo. Nature 2016, 533, 251–254. [Google Scholar] [CrossRef]
- Shahbazi, M.N.; Jedrusik, A.; Vuoristo, S.; Recher, G.; Hupalowska, A.; Bolton, V.; Fogarty, N.M.E.; Campbell, A.; Devito, L.G.; Ilic, D.; et al. Self-organization of the human embryo in the absence of maternal tissues. Nat. Cell Biol. 2016, 18, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, H.; Javali, A.; Khoei, H.H.; Sommer, T.M.; Sestini, G.; Novatchkova, M.; Scholte op Reimer, Y.; Castel, G.; Bruneau, A.; Maenhoudt, N.; et al. Human blastoids model blastocyst development and implantation. Nature 2022, 601, 600–605. [Google Scholar] [CrossRef]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef]
- Huppertz, B. The anatomy of the normal placenta. J. Clin. Pathol. 2008, 61, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jaremek, A.; Jeyarajah, M.J.; Jaju Bhattad, G.; Renaud, S.J. Omics Approaches to Study Formation and Function of Human Placental Syncytiotrophoblast. Front. Cell Dev. Biol. 2021, 9, 674162. [Google Scholar] [CrossRef]
- Soma-Pillay, P.; Nelson-Piercy, C.; Tolppanen, H.; Mebazaa, A. Physiological changes in pregnancy. Cardiovasc. J. Afr. 2016, 27, 89–94. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Sáez, M.A.; Álvarez-Mon, M.A.; Torres-Carranza, D.; Álvarez-Mon, M.; Bujan, J.; García-Honduvilla, N.; Bravo, C.; et al. The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease. Cells 2022, 11, 568. [Google Scholar] [CrossRef]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth-New Insights From Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Balasinor, N.H. Placental Defects: An Epigenetic Perspective. Reprod. Sci. 2018, 25, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, V.; Fineberg, E.; Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J.; et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef]
- Liu, S.; Joseph, K.S.; Lisonkova, S.; Rouleau, J.; Hof, M.V.d.; Sauve, R.; Kramer, M.S. Association between Maternal Chronic Conditions and Congenital Heart Defects. Circulation 2013, 128, 583–589. [Google Scholar] [CrossRef]
- Camm, E.J.; Botting, K.J.; Sferruzzi-Perri, A.N. Near to One’s Heart: The Intimate Relationship Between the Placenta and Fetal Heart. Front. Physiol. 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. 2007, 113, 1–13. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, A.; Xu, H. The roles of vascular endothelial growth factor gene polymorphisms in congenital heart diseases: A meta-analysis. Growth Factors 2018, 36, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Aplin, J.D.; Myers, J.E.; Timms, K.; Westwood, M. Tracking placental development in health and disease. Nat. Rev. Endocrinol. 2020, 16, 479–494. [Google Scholar] [CrossRef]
- Svensson-Arvelund, J.; Mehta, R.B.; Lindau, R.; Mirrasekhian, E.; Rodriguez-Martinez, H.; Berg, G.; Lash, G.E.; Jenmalm, M.C.; Ernerudh, J. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J. Immunol. 2015, 194, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nakashima, A.; Shima, T.; Ito, M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am. J. Reprod. Immunol. 2010, 63, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Carlin, D.; Zhu, F.; Cattaneo, P.; Ideker, T.; Evans, S.M.; Bloomekatz, J.; Chi, N.C. Unveiling Complexity and Multipotentiality of Early Heart Fields. Circ. Res. 2021, 129, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The heart field transcriptional landscape at single-cell resolution. Dev. Cell 2023, 58, 257–266. [Google Scholar] [CrossRef]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Ivanovitch, K.; Soro-Barrio, P.; Chakravarty, P.; Jones, R.A.; Bell, D.M.; Mousavy Gharavy, S.N.; Stamataki, D.; Delile, J.; Smith, J.C.; Briscoe, J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLOS Biol. 2021, 19, e3001200. [Google Scholar] [CrossRef]
- Laugwitz, K.-L.; Moretti, A.; Caron, L.; Nakano, A.; Chien, K.R. Islet1 cardiovascular progenitors: A single source for heart lineages? Development 2008, 135, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.D.; Buckingham, M.E.; Peter, K. How to Make a Heart: The Origin and Regulation of Cardiac Progenitor Cells. Curr. Top Dev. Biol. 2010, 90, 1–41. [Google Scholar] [CrossRef] [PubMed]
- van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Munshi, N.V. Development of the Cardiac Conduction System. Cold Spring Harb. Perspect. Biol. 2020, 12, a037408. [Google Scholar] [CrossRef]
- Mjaatvedt, C.H.; Yamamura, H.; Wessels, A.; Ramsdell, A.; Turner, D.; Markwald, R.R.; Rosenthal, N. 10—Mechanisms of Segmentation, Septation, and Remodeling of the Tubular Heart: Endocardial Cushion Fate and Cardiac Looping A2—Harvey, Richard P. In Heart Development; Academic Press: San Diego, CA, USA, 1999; pp. 159–177. [Google Scholar] [CrossRef]
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef]
- George, R.M.; Maldonado-Velez, G.; Firulli, A.B. The heart of the neural crest: Cardiac neural crest cells in development and regeneration. Development 2020, 147, dev188706. [Google Scholar] [CrossRef] [PubMed]
- Bardot, E.; Calderon, D.; Santoriello, F.; Han, S.; Cheung, K.; Jadhav, B.; Burtscher, I.; Artap, S.; Jain, R.; Epstein, J.; et al. Foxa2 identifies a cardiac progenitor population with ventricular differentiation potential. Nat. Commun. 2017, 8, 14428. [Google Scholar] [CrossRef] [PubMed]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct Compartments of the Proepicardial Organ Give Rise to Coronary Vascular Endothelial Cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef]
- Motoike, T.; Markham, D.W.; Rossant, J.; Sato, T.N. Evidence for novel fate of Flk1+ progenitor: Contribution to muscle lineage. Genesis 2003, 35, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Witman, N.; Zhou, C.; Grote Beverborg, N.; Sahara, M.; Chien, K.R. Cardiac progenitors and paracrine mediators in cardiogenesis and heart regeneration. Semin. Cell Dev. Biol. 2020, 100, 29–51. [Google Scholar] [CrossRef]
- Flaherty, M.P.; Dawn, B. Noncanonical Wnt11 Signaling and Cardiomyogenic Differentiation. Trends Cardiovasc. Med. 2008, 18, 260–268. [Google Scholar] [CrossRef]
- Strate, I.; Tessadori, F.; Bakkers, J. Glypican4 promotes cardiac specification and differentiation by attenuating canonical Wnt and Bmp signaling. Development 2015, 142, 1767–1776. [Google Scholar] [CrossRef]
- Ueno, S.; Weidinger, G.; Osugi, T.; Kohn, A.D.; Golob, J.L.; Pabon, L.; Reinecke, H.; Moon, R.T.; Murry, C.E. Biphasic role for Wnt/β-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9685–9690. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Pfeiffer, M.J.; Frank, S.; Adachi, K.; Piccini, I.; Quaranta, R.; Arauzo-Bravo, M.; Schwarz, J.; Schade, D.; Leidel, S.; et al. Stepwise Clearance of Repressive Roadblocks Drives Cardiac Induction in Human ESCs. Cell Stem Cell 2016, 18, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Buikema, J.W.; Zwetsloot, P.-P.M.; Doevendans, P.A.; Domian, I.J.; PG, S.J. Wnt/β-Catenin Signaling during Cardiac Development and Repair. J. Cardiovasc. Dev. Dis. 2014, 1, 98–110. [Google Scholar] [CrossRef]
- Liu, Y.; Asakura, M.; Inoue, H.; Nakamura, T.; Sano, M.; Niu, Z.; Chen, M.; Schwartz, R.J.; Schneider, M.D. Sox17 is essential for the specification of cardiac mesoderm in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3859–3864. [Google Scholar] [CrossRef]
- Paige, S.L.; Osugi, T.; Afanasiev, O.K.; Pabon, L.; Reinecke, H.; Murry, C.E. Endogenous Wnt/beta-Catenin Signaling Is Required for Cardiac Differentiation in Human Embryonic Stem Cells. PLoS ONE 2010, 5, e11134. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jia, Z.; Wang, T.; Wang, W.; Zhang, C.; Chen, P.; Ma, K.; Zhou, C. Interaction of Wnt/β-catenin and notch signaling in the early stage of cardiac differentiation of P19CL6 cells. J. Cell. Biochem. 2012, 113, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Garside, V.; Chang, A.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Karsan, A. Notch Signaling in Cardiac Development. Circ. Res. 2008, 102, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Urbanek, K.; Nascimbene, A.; Hosoda, T.; Zheng, H.; Delucchi, F.; Amano, K.; Gonzalez, A.; Vitale, S.; Ojaimi, C.; et al. Notch1 regulates the fate of cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 15529–15534. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kokubo, H.; Miyagawa-Tomita, S.; Endo, M.; Igarashi, K.; Aisaki, K.i.; Kanno, J.; Saga, Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development 2006, 133, 1625–1634. [Google Scholar] [CrossRef]
- Shamis, Y.; Cullen, D.; Liu, L.; Yang, G.; Ng, S.; Xiao, L.; Bell, F.; Ray, C.; Takikawa, S.; Moskowitz, I.; et al. Maternal and zygotic Zfp57 modulate NOTCH signaling in cardiac development. Proc. Natl. Acad. Sci. USA 2015, 112, E2020–E2029. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Hrstka, S.C.L.; Evans, J.M.; O’Byrne, M.M.; de Andrade, M.; O’Leary, P.W.; Nelson, T.J.; Olson, T.M. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet. 2015, 134, 1003–1011. [Google Scholar] [CrossRef]
- Luxan, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- D’Amato, G.; Luxán, G.; de la Pompa, J.L. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J. 2016, 283, 4223–4237. [Google Scholar] [CrossRef]
- MacGrogan, D.; Luxán, G.; de la Pompa, J.L. Genetic and functional genomics approaches targeting the Notch pathway in cardiac development and congenital heart disease. Brief. Funct. Genom. 2014, 13, 15–27. [Google Scholar] [CrossRef]
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef]
- Miao, L.; Lu, Y.; Nusrat, A.; Abdelnasser, H.Y.; Datta, S.; Zhou, B.; Schwartz, R.J.; Wu, M. The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis. Cells 2021, 10, 2192. [Google Scholar] [CrossRef]
- del Monte-Nieto, G.; Ramialison, M.; Adam, A.A.S.; Wu, B.; Aharonov, A.; D’Uva, G.; Bourke, L.M.; Pitulescu, M.E.; Chen, H.; de la Pompa, J.L.; et al. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 2018, 557, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.J.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2-5/Bmp2/Smad1 Negative Feedback Loop Controls Heart Progenitor Specification and Proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Greene, S.B.; Bonilla-Claudio, M.; Tao, Y.; Zhang, J.; Bai, Y.; Huang, Z.; Black, B.L.; Wang, F.; Martin, J.F. Bmp Signaling Regulates Myocardial Differentiation from Cardiac Progenitors through a MicroRNA-Mediated Mechanism. Dev. Cell 2010, 19, 903–912. [Google Scholar] [CrossRef]
- van Wijk, B.; Moorman, A.F.M.; van den Hoff, M.J.B. Role of bone morphogenetic proteins in cardiac differentiation. Cardiovasc. Res. 2007, 74, 244–255. [Google Scholar] [CrossRef]
- Yuasa, S.; Fukuda, K. Multiple roles for BMP signaling in cardiac development. Drug Discov. Today Ther. Strateg. 2008, 5, 209–214. [Google Scholar] [CrossRef]
- McCulley, D.J.; Kang, J.-O.; Martin, J.F.; Black, B.L. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 2008, 237, 3200–3209. [Google Scholar] [CrossRef]
- Cai, W.; Albini, S.; Wei, K.; Willems, E.; Guzzo, R.M.; Tsuda, M.; Giordani, L.; Spiering, S.; Kurian, L.; Yeo, G.W.; et al. Coordinate Nodal and BMP inhibition directs Baf60c-dependent cardiomyocyte commitment. Genes Dev. 2013, 27, 2332–2344. [Google Scholar] [CrossRef]
- Taha, M.F.; Valojerdi, M.R. Effect of bone morphogenetic protein-4 on cardiac differentiation from mouse embryonic stem cells in serum-free and low-serum media. Int. J. Cardiol. 2008, 127, 78–87. [Google Scholar] [CrossRef]
- Tirosh-Finkel, L.; Zeisel, A.; Brodt-Ivenshitz, M.; Shamai, A.; Yao, Z.; Seger, R.; Domany, E.; Tzahor, E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 2010, 137, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; McColl, J.; Camp, E.; Kennerley, N.; Mok, G.F.; McCormick, D.; Grocott, T.; Wheeler, G.N.; Münsterberg, A.E. Smad1 transcription factor integrates BMP2 and Wnt3a signals in migrating cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 7337–7342. [Google Scholar] [CrossRef]
- Luo, W.; Zhao, X.; Jin, H.; Tao, L.; Zhu, J.; Wang, H.; Hemmings, B.A.; Yang, Z. Akt1 signaling coordinates BMP signaling and β-catenin activity to regulate second heart field progenitor development. Development 2015, 142, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ohta, H.; Nakayama, Y.; Konishi, M. Roles of FGF Signals in Heart Development, Health, and Disease. Front. Cell Dev. Biol. 2016, 4, 110. [Google Scholar] [CrossRef] [PubMed]
- Hubert, F.; Payan, S.M.; Rochais, F. FGF10 Signaling in Heart Development, Homeostasis, Disease and Repair. Front. Genet. 2018, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Floriddia, E.M.; Lourenço, T.; Zhang, S.; van Bruggen, D.; Hilscher, M.M.; Kukanja, P.; Gonçalves dos Santos, J.P.; Altınkök, M.; Yokota, C.; Llorens-Bobadilla, E.; et al. Distinct oligodendrocyte populations have spatial preference and different responses to spinal cord injury. Nat. Commun. 2020, 11, 5860. [Google Scholar] [CrossRef] [PubMed]
- Engels, M.C.; Rajarajan, K.; Feistritzer, R.; Sharma, A.; Nielsen, U.B.; Schalij, M.J.; de Vries, A.A.F.; Pijnappels, D.A.; Wu, S.M. Insulin-Like Growth Factor Promotes Cardiac Lineage Induction In Vitro by Selective Expansion of Early Mesoderm. Stem Cells 2014, 32, 1493–1502. [Google Scholar] [CrossRef]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.; Hughes, J.; Hassan, A.B.; Brüning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef]
- Wang, H.; Xie, Y.; Guan, L.; Elkin, K.; Xiao, J. Targets identified from exercised heart: Killing multiple birds with one stone. NPJ Regen. Med. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.L.; Tham, Y.K.; Yildiz, S.G.; Alexander, Y.; Donner, D.G.; Kiriazis, H.; Harmawan, C.A.; Hsu, A.; Bernardo, B.C.; Matsumoto, A.; et al. FoxO1 is required for physiological cardiac hypertrophy induced by exercise but not by constitutively active PI3K. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H1470–H1485. [Google Scholar] [CrossRef] [PubMed]
- Bass-Stringer, S.; Ooi, J.Y.Y.; McMullen, J.R. Clusterin is regulated by IGF1–PI3K signaling in the heart: Implications for biomarker and drug target discovery, and cardiotoxicity. Arch. Toxicol. 2020, 94, 1763–1768. [Google Scholar] [CrossRef]
- Wederell, E.D.; Bilenky, M.; Cullum, R.; Thiessen, N.; Dagpinar, M.; Delaney, A.; Varhol, R.; Zhao, Y.; Zeng, T.; Bernier, B.; et al. Global analysis of in vivo Foxa2-binding sites in mouse adult liver using massively parallel sequencing. Nucl. Acids Res. 2008, 36, 4549–4564. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Grunewald, M.; Dor, Y.; Keshet, E. VEGF regulates relative allocation of Isl1+ cardiac progenitors to myocardial and endocardial lineages. Mech. Dev. 2016, 142, 40–49. [Google Scholar] [CrossRef]
- Dor, Y.; Camenisch, T.D.; Itin, A.; Fishman, G.I.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-H.; Gehmert, S.; Sadat, S.; Pinkernell, K.; Bai, X.; Matthias, N.; Alt, E. VEGF is critical for spontaneous differentiation of stem cells into cardiomyocytes. Biochem. Biophys. Res. Commun. 2007, 354, 999–1003. [Google Scholar] [CrossRef]
- Lania, G.; Ferrentino, R.; Baldini, A. TBX1 Represses Vegfr2 Gene Expression and Enhances the Cardiac Fate of VEGFR2+ Cells. PLoS ONE 2015, 10, e0138525. [Google Scholar] [CrossRef]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the Tgf-[beta] type I receptor Alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef]
- Duelen, R.; Gilbert, G.; Patel, A.; de Schaetzen, N.; De Waele, L.; Roderick, L.; Sipido, K.R.; Verfaillie, C.M.; Buyse, G.M.; Thorrez, L.; et al. Activin A Modulates CRIPTO-1/HNF4α+ Cells to Guide Cardiac Differentiation from Human Embryonic Stem Cells. Stem Cells Int. 2017, 2017, 4651238. [Google Scholar] [CrossRef]
- Deshwar, A.R.; Chng, S.C.; Ho, L.; Reversade, B.; Scott, I.C. The Apelin receptor enhances Nodal/TGFβ signaling to ensure proper cardiac development. eLife 2016, 5, e13758. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Haase, A.; Franke, A.; Bolesani, E.; Schwanke, K.; Robles-Diaz, D.; Coffee, M.; Göhring, G.; Dräger, G.; et al. Bulk cell density and Wnt/TGFbeta signalling regulate mesendodermal patterning of human pluripotent stem cells. Nat. Commun. 2016, 7, 13602. [Google Scholar] [CrossRef]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Cohen, E.D.; Morrisey, E.E. The Importance of Wnt Signaling in Cardiovascular Development. Pediatr. Cardiol. 2010, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Freire, A.G.; Resende, T.P.; Pinto-do-Ó, P. Building and Repairing the Heart: What Can We Learn from Embryonic Development? BioMed Res. Int. 2014, 2014, 679168. [Google Scholar] [CrossRef] [PubMed]
- Onizuka, T.; Yuasa, S.; Kusumoto, D.; Shimoji, K.; Egashira, T.; Ohno, Y.; Kageyama, T.; Tanaka, T.; Hattori, F.; Fujita, J.; et al. Wnt2 accelerates cardiac myocyte differentiation from ES-cell derived mesodermal cells via non-canonical pathway. J. Mol. Cell. Cardiol. 2012, 52, 650–659. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Li, Y.; Qian, H.; Shan, X.; Yan, Y.; Mao, F.; Wu, X.; Xu, W.R. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle 2011, 10, 3198–3207. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine Mechanisms in Adult Stem Cell Signaling and Therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef] [PubMed]
- Rafatian, G.; Davis, D.R. Concise Review: Heart-Derived Cell Therapy 2.0: Paracrine Strategies to Increase Therapeutic Repair of Injured Myocardium. Stem Cells 2018, 36, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of Angiogenic and Antiapoptotic Factors by Human Adipose Stromal Cells. Circulation 2004, 109, 1292–1298. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.E.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteom. 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Liu, C.; Bayado, N.; He, D.; Li, J.; Chen, H.; Li, L.; Li, J.; Long, X.; Du, T.; Tang, J.; et al. Therapeutic Applications of Extracellular Vesicles for Myocardial Repair. Front. Cardiovasc. Med. 2021, 8, 758050. [Google Scholar] [CrossRef]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W.; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma Exosomes Protect the Myocardium From Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef]
- Li, S.; Jin, Y.; Tang, P.; Liu, X.; Chai, X.; Dong, J.; Che, X.; Zhou, Q.; Ni, M.; Jin, F. Maternal serum-derived exosomal lactoferrin as a marker in detecting and predicting ventricular septal defect in fetuses. Exp. Biol. Med. 2022, 247, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Shen, H.; Wu, S.; Ruan, J.; Lyu, G. Construction of the amniotic fluid-derived exosomal ceRNA network associated with ventricular septal defect. Genomics 2021, 113, 4293–4302. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.-h.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef]
- Loyer, X.; Vion, A.-C.; Tedgui, A.; Boulanger, C.M. Microvesicles as Cell–Cell Messengers in Cardiovascular Diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef]
- Wang, X.; Huang, W.; Liu, G.; Cai, W.; Millard, R.W.; Wang, Y.; Chang, J.; Peng, T.; Fan, G.-C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014, 74, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, Q. Potential clinical applications of exosomes in the diagnosis, treatment, and prognosis of cardiovascular diseases: A narrative review. Ann. Transl. Med. 2022, 10, 372. [Google Scholar] [CrossRef]
- Kishore, R.; Khan, M. Cardiac cell-derived exosomes: Changing face of regenerative biology. Eur. Heart J. 2016, 38, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Ghafourian, M.; Mahdavi, R.; Akbari Jonoush, Z.; Sadeghi, M.; Ghadiri, N.; Farzaneh, M.; Mousavi Salehi, A. The implications of exosomes in pregnancy: Emerging as new diagnostic markers and therapeutics targets. Cell Commun. Signal. 2022, 20, 51. [Google Scholar] [CrossRef]
- Sarker, S.; Scholz-Romero, K.; Perez, A.; Illanes, S.E.; Mitchell, M.D.; Rice, G.E.; Salomon, C. Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy. J. Transl. Med. 2014, 12, 204. [Google Scholar] [CrossRef]
- Paktinat, S.; Hashemi, S.M.; Ghaffari Novin, M.; Mohammadi-Yeganeh, S.; Salehpour, S.; Karamian, A.; Nazarian, H. Seminal exosomes induce interleukin-6 and interleukin-8 secretion by human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 235, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Greening, D.W.; Catt, S.; Salamonsen, L.; Evans, J. Exosomes and soluble secretome from hormone-treated endometrial epithelial cells direct embryo implantation. Mol. Hum. Reprod. 2020, 26, 510–520. [Google Scholar] [CrossRef]
- Shi, R.; Zhao, L.; Cai, W.; Wei, M.; Zhou, X.; Yang, G.; Yuan, L. Maternal exosomes in diabetes contribute to the cardiac development deficiency. Biochem. Biophys. Res. Commun. 2017, 483, 602–608. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wang, C.; Shi, R.; Zhou, X.; Li, Z.; Sun, W.; Zhao, L.; Yuan, L. Maternal obesity increases the risk of fetal cardiac dysfunction via visceral adipose tissue derived exosomes. Placenta 2021, 105, 85–93. [Google Scholar] [CrossRef]
- Salomon, C.; Guanzon, D.; Scholz-Romero, K.; Longo, S.; Correa, P.; Illanes, S.E.; Rice, G.E. Placental Exosomes as Early Biomarker of Preeclampsia: Potential Role of Exosomal MicroRNAs Across Gestation. J. Clin. Endocrinol. Metab. 2017, 102, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Burkova, E.E.; Sedykh, S.E.; Nevinsky, G.A. Human Placenta Exosomes: Biogenesis, Isolation, Composition, and Prospects for Use in Diagnostics. Int. J. Mol. Sci. 2021, 22, 2158. [Google Scholar] [CrossRef]
- Jin, J.; Menon, R. Placental exosomes: A proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 2018, 79, e12788. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Barnes, R.M.; Black, B.L. Nodal Signaling and Congenital Heart Defects. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer Japan: Tokyo, Japan, 2016; pp. 183–192. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. Activin/Nodal signalling in stem cells. Development 2015, 142, 607–619. [Google Scholar] [CrossRef]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef]
- Liu, L.; Nemashkalo, A.; Rezende, L.; Jung, J.Y.; Chhabra, S.; Guerra, M.C.; Heemskerk, I.; Warmflash, A. Nodal is a short-range morphogen with activity that spreads through a relay mechanism in human gastruloids. Nat. Commun. 2022, 13, 497. [Google Scholar] [CrossRef]
- Webber, D.M.; Li, M.; MacLeod, S.L.; Tang, X.; Levy, J.W.; Karim, M.A.; Erickson, S.W.; Hobbs, C.A.; The National Birth Defects Prevention Study. Gene-Folic Acid Interactions and Risk of Conotruncal Heart Defects: Results from the National Birth Defects Prevention Study. Genes 2023, 14, 180. [Google Scholar] [CrossRef]
- Wondemagegn, A.T.; Afework, M. The association between folic acid supplementation and congenital heart defects: Systematic review and meta-analysis. SAGE Open Med. 2022, 10, 20503121221081069. [Google Scholar] [CrossRef]
- Kumar, S.D.; Vijaya, M.; Samy, R.P.; Dheen, S.T.; Ren, M.; Watt, F.; Kang, Y.J.; Bay, B.-H.; Tay, S.S.W. Zinc supplementation prevents cardiomyocyte apoptosis and congenital heart defects in embryos of diabetic mice. Free. Radic. Biol. Med. 2012, 53, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Vermot, J.; Messaddeq, N.; Niederreither, K.; Dierich, A.; Dollé, P. Rescue of morphogenetic defects and of retinoic acid signaling in retinaldehyde dehydrogenase 2 (Raldh2) mouse mutants by chimerism with wild-type cells. Differentiation 2006, 74, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Chiş, A.R.; Moise, A.R. Role of carotenoids and retinoids during heart development. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158636. [Google Scholar] [CrossRef] [PubMed]
- Agnew, E.J.; Ivy, J.R.; Stock, S.J.; Chapman, K.E. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J. Mol. Endocrinol. 2018, 61, R61–R73. [Google Scholar] [CrossRef]
- Breur, J.M.; Visser, G.H.; Kruize, A.A.; Stoutenbeek, P.; Meijboom, E.J. Treatment of fetal heart block with maternal steroid therapy: Case report and review of the literature. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2004, 24, 467–472. [Google Scholar] [CrossRef]
- Eliasson, H.; Sonesson, S.E.; Sharland, G.; Granath, F.; Simpson, J.M.; Carvalho, J.S.; Jicinska, H.; Tomek, V.; Dangel, J.; Zielinsky, P.; et al. Isolated atrioventricular block in the fetus: A retrospective, multinational, multicenter study of 175 patients. Circulation 2011, 124, 1919–1926. [Google Scholar] [CrossRef]
- Gaber, N.; Gagliardi, M.; Patel, P.; Kinnear, C.; Zhang, C.; Chitayat, D.; Shannon, P.; Jaeggi, E.; Tabori, U.; Keller, G.; et al. Fetal Reprogramming and Senescence in Hypoplastic Left Heart Syndrome and in Human Pluripotent Stem Cells during Cardiac Differentiation. Am. J. Pathol. 2013, 183, 720–734. [Google Scholar] [CrossRef]
- Harding, J.; Bauer, M.; Kimble, R. Antenatal therapy for intrauterine growth retardation. Acta Paediatr. Suppl. 1997, 86, 196–200. [Google Scholar] [CrossRef]
- Laviola, L.; Perrini, S.; Belsanti, G.; Natalicchio, A.; Montrone, C.; Leonardini, A.; Vimercati, A.; Scioscia, M.; Selvaggi, L.; Giorgino, R.; et al. Intrauterine Growth Restriction in Humans Is Associated with Abnormalities in Placental Insulin-Like Growth Factor Signaling. Endocrinology 2005, 146, 1498–1505. [Google Scholar] [CrossRef]
- Marbán, E. A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat. Biomed. Eng. 2018, 2, 353–361. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Lässer, C.; Eldh, M.; Lötvall, J. Isolation and Characterization of RNA-Containing Exosomes. JoVE 2012, 59, e3037. [Google Scholar] [CrossRef]
- Rosso, G.; Cauda, V. Biomimicking Extracellular Vesicles with Fully Artificial Ones: A Rational Design of EV-BIOMIMETICS toward Effective Theranostic Tools in Nanomedicine. ACS Biomater. Sci. Eng. 2022. [Google Scholar] [CrossRef]
- Cao, S.; Wu, Y.; Albert Reece, E.; Xu, C.; Shen, W.-B.; Kaushal, S.; Yang, P. Functional cargos of exosomes derived from Flk-1+ vascular progenitors enable neurulation and ameliorate embryonic anomalies in diabetic pregnancy. Commun. Biol. 2022, 5, 648. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zhao, Q.; Beck, A.; Vitale, J.M.; Schneider, J.S.; Terzic, A.; Fraidenraich, D. Rescue of Developmental Defects by Blastocyst Stem Cell Injection: Towards Elucidation of Neomorphic Corrective Pathways. J. Cardiovasc. Transl. Res. 2010, 3, 66–72. [Google Scholar] [CrossRef]
- Fraidenraich, D.; Stillwell, E.; Romero, E.; Wilkes, D.; Manova, K.; Basson, C.; Benezra, R. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science 2004, 306, 247–252. [Google Scholar] [CrossRef]
- Santos, J.M.A.; Mendes-Silva, L.; Afonso, V.; Martins, G.; Machado, R.S.R.; Lopes, J.A.; Cancela, L.; Futschik, M.E.; Sachinidis, A.; Gavaia, P.; et al. Exogenous Wnt5a and Wnt11 proteins rescue Cited2 dysfunction in mouse embryonic stem cells and zebrafish morphants. Cell Death Dis. 2019, 10, 582. [Google Scholar] [CrossRef] [PubMed]
- Bragança, J.; Mendes-Silva, L.; Lopes, J.; SM, C. CITED proteins in the heart of pluripotent cells and in heart’s full potential. Regen Med. Front. 2019, 1, e190005. [Google Scholar] [CrossRef]
- Pacheco-Leyva, I.; Matias, A.C.; Oliveira, D.V.; Santos, J.M.A.; Nascimento, R.; Guerreiro, E.; Michell, A.C.; van De Vrugt, A.M.; Machado-Oliveira, G.; Ferreira, G.; et al. CITED2 Cooperates with ISL1 and Promotes Cardiac Differentiation of Mouse Embryonic Stem Cells. Stem Cell Rep. 2016, 7, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, S.T.; Bamforth, S.D.; Bragança, J.; Chen, C.-M.; Broadbent, C.; Schneider, J.E.; Schwartz, R.J.; Bhattacharya, S. A cell-autonomous role of Cited2 in controlling myocardial and coronary vascular development. Eur. Heart J. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-m.; Bentham, J.; Cosgrove, C.; Braganca, J.; Cuenda, A.; Bamforth, S.D.; Schneider, J.E.; Watkins, H.; Keavney, B.; Davies, B.; et al. Functional Significance of SRJ Domain Mutations in CITED2. PLoS ONE 2012, 7, e46256. [Google Scholar] [CrossRef]
- MacDonald, S.T.; Bamforth, S.D.; Chen, C.-M.; Farthing, C.R.; Franklyn, A.; Broadbent, C.; Schneider, J.E.; Saga, Y.; Lewandoski, M.; Bhattacharya, S. Epiblastic Cited2 deficiency results in cardiac phenotypic heterogeneity and provides a mechanism for haploinsufficiency. Cardiovasc. Res. 2008, 79, 448–457. [Google Scholar] [CrossRef]
- Bamforth, S.D.; Bragança, J.; Farthing, C.R.; Schneider, J.E.; Broadbent, C.; Michell, A.C.; Clarke, K.; Neubauer, S.; Norris, D.; Brown, N.A.; et al. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 2004, 36, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Bragança, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.R.; Kranc, K.R.; Farza, H.; Henderson, D.J.; Hurst, H.C.; Bhattacharya, S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef]
- Xu, B.; Doughman, Y.; Turakhia, M.; Jiang, W.; Landsettle, C.E.; Agani, F.H.; Semenza, G.L.; Watanabe, M.; Yang, Y.-C. Partial rescue of defects in Cited2-deficient embryos by HIF-1[alpha] heterozygosity. Dev. Biol. 2007, 301, 130. [Google Scholar] [CrossRef][Green Version]
- Kranc, K.R.; Oliveira, D.V.; Armesilla-Diaz, A.; Pacheco-Leyva, I.; Matias, A.C.; Escapa, A.L.; Subramani, C.; Wheadon, H.; Trindade, M.; Nichols, J.; et al. Acute loss of Cited2 impairs Nanog expression and decreases self-renewal of mouse embryonic stem cells. Stem Cells 2015, 33, 699–712. [Google Scholar] [CrossRef]
- Webber, S.A.; Wargowski, D.S.; Chitayat, D.; Sandor, G.G.S. Congenital heart disease and Robinow syndrome: Coincidence or an additional component of the syndrome? Am. J. Med. Genet. 1990, 37, 519–521. [Google Scholar] [CrossRef]
- Li, P.; Li, H.; Zheng, Y.; Qiao, B.; Duan, W.; Huang, L.; Liu, W.; Wang, H. Variants in the Regulatory Region of WNT5A Reduced Risk of Cardiac Conotruncal Malformations in the Chinese Population. Sci. Rep. 2015, 5, 13120. [Google Scholar] [CrossRef]
- Škorić-Milosavljević, D.; Tadros, R.; Bosada, F.M.; Tessadori, F.; Weerd, J.H.v.; Woudstra, O.I.; Tjong, F.V.Y.; Lahrouchi, N.; Bajolle, F.; Cordell, H.J.; et al. Common Genetic Variants Contribute to Risk of Transposition of the Great Arteries. Circ. Res. 2022, 130, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Li, D.; Théveniau-Ruissy, M.; Hutson, M.R.; Kelly, R.G.; Wang, J. Loss of Wnt5a disrupts second heart field cell deployment and may contribute to OFT malformations in DiGeorge syndrome. Hum. Mol. Genet. 2015, 24, 1704–1716. [Google Scholar] [CrossRef]
- Yu, H.; Ye, X.; Guo, N.; Nathans, J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: Evidence for a network of interacting genes. Development 2012, 139, 4383–4394. [Google Scholar] [CrossRef]
- Schleiffarth, J.R.; Person, A.D.; Martinsen, B.J.; Sukovich, D.J.; Neumann, A.; Baker, C.V.H.; Lohr, J.L.; Cornfield, D.N.; Ekker, S.C.; Petryk, A. Wnt5a Is Required for Cardiac Outflow Tract Septation in Mice. Pediatr. Res. 2007, 61, 386. [Google Scholar] [CrossRef]
- Zhao, Y.; Kang, X.; Gao, F.; Guzman, A.; Lau, R.P.; Biniwale, R.; Wadehra, M.; Reemtsen, B.; Garg, M.; Halnon, N.; et al. Gene-environment regulatory circuits of right ventricular pathology in tetralogy of fallot. J. Mol. Med. 2019, 97, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Erikson, D.W.; Tilford, S.A.; Bany, B.M.; Maclean, I.I.J.A.; Rucker, I.I.I.E.B.; Johnson, G.A.; Spencer, T.E. Wnt Genes in the Mouse Uterus: Potential Regulation of Implantation. Biol. Reprod. 2009, 80, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, G.; Saleh, L.; Otti, G.R.; Haider, S.; Velicky, P.; Fiala, C.; Pollheimer, J.r.; Knöfler, M. Wingless ligand 5a is a critical regulator of placental growth and survival. Sci. Rep. 2016, 6, 28127. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef]
- Sinha, T.; Lin, L.; Li, D.; Davis, J.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Mapping the dynamic expression of Wnt11 and the lineage contribution of Wnt11-expressing cells during early mouse development. Dev. Biol. 2015, 398, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Bisson, J.A.; Mills, B.; Paul Helt, J.-C.; Zwaka, T.P.; Cohen, E.D. Wnt5a and Wnt11 inhibit the canonical Wnt pathway and promote cardiac progenitor development via the Caspase-dependent degradation of AKT. Dev. Biol. 2015, 398, 80–96. [Google Scholar] [CrossRef]
- Cohen, E.D.; Miller, M.F.; Wang, Z.; Moon, R.T.; Morrisey, E.E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012, 139, 1931–1940. [Google Scholar] [CrossRef]
- Chen, M.; Qian, C.; Bi, L.-L.; Zhao, F.; Zhang, G.-Y.; Wang, Z.-Q.; Gan, X.-D.; Wang, Y.-G. Enrichment of cardiac differentiation by a large starting number of embryonic stem cells in embryoid bodies is mediated by the Wnt11-JNK pathway. Biotechnol. Lett. 2015, 37, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.M.; Garriock, R.J.; Yatskievych, T.A.; D’Agostino, S.L.; Antin, P.B.; Krieg, P.A. Non-canonical Wnt signaling through Wnt5a/b and a novel Wnt11 gene, Wnt11b, regulates cell migration during avian gastrulation. Dev. Biol. 2008, 320, 391–401. [Google Scholar] [CrossRef]
- Vijayaragavan, K.; Szabo, E.; Bossé, M.; Ramos-Mejia, V.; Moon, R.T.; Bhatia, M. Noncanonical Wnt Signaling Orchestrates Early Developmental Events toward Hematopoietic Cell Fate from Human Embryonic Stem Cells. Cell Stem Cell 2009, 4, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Mastelaro de Rezende, M.; Zenker Justo, G.; Julian Paredes-Gamero, E.; Gosens, R. Wnt-5A/B Signaling in Hematopoiesis throughout Life. Cells 2020, 9, 1801. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, A.; Choi, S.Y.; Kan, Q.; Zhou, W.; Chacon-Heszele, M.F.; Ryu, Y.K.; McKenna, S.; Zuo, X.; Kuruvilla, R.; et al. Wnt5a Is Necessary for Normal Kidney Development in Zebrafish and Mice. Nephron Exp. Nephrol. 2014, 128, 80–88. [Google Scholar] [CrossRef]
- Ryu, Y.K.; Collins, S.E.; Ho, H.-Y.H.; Zhao, H.; Kuruvilla, R. An autocrine Wnt5a-Ror signaling loop mediates sympathetic target innervation. Dev. Biol. 2013, 377, 79–89. [Google Scholar] [CrossRef]
- Yang, R.; Goedel, A.; Kang, Y.; Si, C.; Chu, C.; Zheng, Y.; Chen, Z.; Gruber, P.J.; Xiao, Y.; Zhou, C.; et al. Amnion signals are essential for mesoderm formation in primates. Nat. Commun. 2021, 12, 5126. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Lanza, R. Next-generation stem cells—Ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 2020, 19, 463–479. [Google Scholar] [CrossRef]
- Jeensuk, S.; Ortega, M.S.; Saleem, M.; Hawryluk, B.; Scheffler, T.L.; Hansen, P.J. Actions of WNT family member 5A to regulate characteristics of development of the bovine preimplantation embryo. Biol. Reprod. 2022, 107, 928–944. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Bartos, A.; Park, C.; Sun, X.; Li, Y.; Cha, S.-W.; Ajima, R.; Ho, H.-Y.H.; Yamaguchi, T.P.; Dey, S.K. Appropriate crypt formation in the uterus for embryo homing and implantation requires Wnt5a-ROR signaling. Cell Rep. 2014, 8, 382–392. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Z.; Chen, C.; Zhang, Y.; Zhang, C. Dysfunction of WNT4/WNT5A in deciduas: Possible relevance to the pathogenesis of preeclampsia. J. Hypertens. 2016, 34, 719–727. [Google Scholar] [CrossRef]
- Dietrich, B.; Haider, S.; Meinhardt, G.; Pollheimer, J.; Knöfler, M. WNT and NOTCH signaling in human trophoblast development and differentiation. Cell. Mol. Life Sci. 2022, 79, 292. [Google Scholar] [CrossRef] [PubMed]
- Rider, V.; Talbott, A.; Bhusri, A.; Krumsick, Z.; Foster, S.; Wormington, J.; Kimler, B.F. WINGLESS (WNT) signaling is a progesterone target for rat uterine stromal cell proliferation. J. Endocrinol. 2016, 229, 197–207. [Google Scholar] [CrossRef]
- Cha, S.-W.; Tadjuidje, E.; White, J.; Wells, J.; Mayhew, C.; Wylie, C.; Heasman, J. Wnt11/5a Complex Formation Caused by Tyrosine Sulfation Increases Canonical Signaling Activity. Curr. Biol. 2009, 19, 1573–1580. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Gold, J.; Xu, C.; Hassanipour, M.; Rosler, E.; Police, S.; Muskheli, V.; Murry, C.E. Formation of Human Myocardium in the Rat Heart from Human Embryonic Stem Cells. Am. J. Pathol. 2005, 167, 663–671. [Google Scholar] [CrossRef]
- van Laake, L.W.; Passier, R.; Monshouwer-Kloots, J.; Verkleij, A.J.; Lips, D.J.; Freund, C.; den Ouden, K.; Ward-van Oostwaard, D.; Korving, J.; Tertoolen, L.G.; et al. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007, 1, 9–24. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef]
- Van den Eynde, J.; Manlhiot, C.; Van De Bruaene, A.; Diller, G.P.; Frangi, A.F.; Budts, W.; Kutty, S. Medicine-Based Evidence in Congenital Heart Disease: How Artificial Intelligence Can Guide Treatment Decisions for Individual Patients. Front. Cardiovasc. Med. 2021, 8, 798215. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, Y.; Sun, L.; Sun, X.; Zhao, X.; Sun, X.; Qian, H.; Xu, W.; Zhu, W. Exosomes Derived from Akt-Modified Human Umbilical Cord Mesenchymal Stem Cells Improve Cardiac Regeneration and Promote Angiogenesis via Activating Platelet-Derived Growth Factor D. Stem Cells Transl. Med. 2017, 6, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P. Cardiac cell therapy: Current status, challenges and perspectives. Arch. Cardiovasc. Dis. 2020, 113, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Balbi, C.; Vassalli, G. Exosomes: Beyond stem cells for cardiac protection and repair. Stem Cells 2020, 38, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Abraham, M.R.; Van Eyk, J.E. Cardiac stem/progenitor cells, secreted proteins, and proteomics. FEBS Lett. 2009, 583, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.; Knoepfler, P. Selling Stem Cells in the USA: Assessing the Direct-to-Consumer Industry. Cell Stem Cell 2016, 19, 154–157. [Google Scholar] [CrossRef]
- Aly, R.M. Current state of stem cell-based therapies: An overview. Stem Cell Investig. 2020, 7, 8. [Google Scholar] [CrossRef]
- Zarzeczny, A.; Atkins, H.; Illes, J.; Kimmelman, J.; Master, Z.; Robillard, J.M.; Snyder, J.; Turner, L.; Zettler, P.J.; Caulfield, T. The stem cell market and policy options: A call for clarity. J. Law Biosci. 2018, 5, 743–758. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).