
Identification of a Novel Variant in Myelin Regulatory Growth Factor by Next-Generation Sequencing Led to the Detection of a Clinically Inapparent Congenital Heart Defect in a Patient with a 46,XY Disorder of Sex Development

 , ,
, , 
Abstract
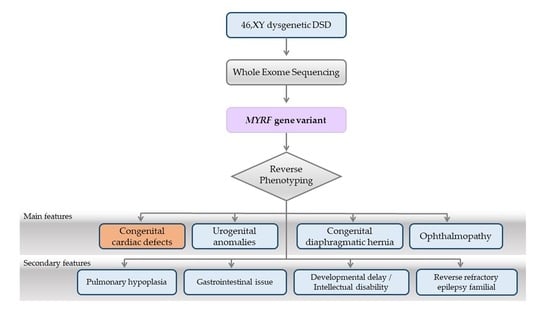
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Clinical Assessment and Diagnostic Testing
2.2. Next Generation Sequencing (NGS) and Filtering
2.3. Sanger Sequencing
3. Results
3.1. Clinical Observations and Diagnostic Testing
3.2. Variant Filtering and Prioritisation
3.3. Reverse Phenotyping
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grinspon, R.P.; Bergadá, I.; Rey, R.A. Male Hypogonadism and Disorders of Sex Development. Front. Endocrinol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A. In collaboration with the participants in the International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus Statement on Management of Intersex Disorders. Pediatrics 2006, 118, e488–e500. [Google Scholar] [CrossRef] [PubMed]
- Globa, E.; Zelinska, N.; Shcherbak, Y.; Bignon-Topalovic, J.; Bashamboo, A.; McElreavey, K. Disorders of Sex Development in a Large Ukrainian Cohort: Clinical Diversity and Genetic Findings. Front. Endocrinol. 2022, 13, 810782. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; Batista, R.L.; Nishi, M.Y.; Lerario, A.M.; Silva, T.E.; de Moraes Narcizo, A.; Benedetti, A.F.F.; de Assis Funari, M.F.; Faria Junior, J.A.; Moraes, D.R.; et al. Contribution of Clinical and Genetic Approaches for Diagnosing 209 Index Cases With 46,XY Differences of Sex Development. J. Clin. Endocrinol. Metab. 2022, 107, e1797–e1806. [Google Scholar] [CrossRef]
- Delot, E.C.; Vilain, E. Towards improved genetic diagnosis of human differences of sex development. Nat. Rev. Genet. 2021, 22, 588–602. [Google Scholar] [CrossRef]
- Lejarraga, H.; del Pino, M.; Fano, V.; Caino, S.; Cole, T.J. Growth references for weight and height for Argentinian girls and boys from birth to maturity: Incorporation of data from the World Health Organisation from birth to 2 years and calculation of new percentiles and LMS values. Arch. Argent. Pediatr. 2009, 107, 126–133. [Google Scholar]
- van der Straaten, S.; Springer, A.; Zecic, A.; Hebenstreit, D.; Tonnhofer, U.; Gawlik, A.; Baumert, M.; Szeliga, K.; Debulpaep, S.; Desloovere, A.; et al. The External Genitalia Score (EGS): A European Multicenter Validation Study. J. Clin. Endocrinol. Metab. 2020, 105, e222–e230. [Google Scholar] [CrossRef]
- Bergadá, I.; Milani, C.; Bedecarrás, P.; Andreone, L.; Ropelato, M.G.; Gottlieb, S.; Bergadá, C.; Campo, S.; Rey, R.A. Time course of the serum gonadotropin surge, inhibins, and anti-Mullerian hormone in normal newborn males during the first month of life. J. Clin. Endocrinol. Metab. 2006, 91, 4092–4098. [Google Scholar] [CrossRef]
- Ballerini, M.G.; Chiesa, A.; Scaglia, P.; Gruneiro-Papendieck, L.; Heinrich, J.J.; Ropelato, M.G. 17alpha-hydroxyprogesterone and cortisol serum levels in neonates and young children: Influence of age, gestational age, gender and methodological procedures. J. Pediatr. Endocrinol. Metab. 2010, 23, 121–132. [Google Scholar] [CrossRef]
- Ballerini, M.G.; Gaido, V.; Rodriguez, M.E.; Chiesa, A.; Ropelato, M.G. Prospective and Descriptive Study on Serum Androstenedione Concentration in Healthy Children from Birth until 18 Years of Age and Its Associated Factors. Dis. Markers 2017, 2017, 9238304. [Google Scholar] [CrossRef]
- Grinspon, R.P.; Nevado, J.; Mori Alvarez, M.L.; del Rey, G.; Castera, R.; Venara, M.; Chiesa, A.; Podestá, M.; Lapunzina, P.; Rey, R.A. 46,XX ovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin. Endocrinol. 2016, 85, 669–675. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Lefter, M.; Vis, J.K.; Vermaat, M.; den Dunnen, J.T.; Taschner, P.E.M.; Laros, J.F.J. Mutalyzer 2: Next generation HGVS nomenclature checker. Bioinformatics 2021, 37, 2811–2817. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef]
- Huang, H.; Zhou, F.; Zhou, S.; Qiu, M. MYRF: A Mysterious Membrane-Bound Transcription Factor Involved in Myelin Development and Human Diseases. Neurosci. Bull. 2021, 37, 881–884. [Google Scholar] [CrossRef]
- Bujalka, H.; Koenning, M.; Jackson, S.; Perreau, V.M.; Pope, B.; Hay, C.M.; Mitew, S.; Hill, A.F.; Lu, Q.R.; Wegner, M.; et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol. 2013, 11, e1001625. [Google Scholar] [CrossRef]
- Li, Z.; Park, Y.; Marcotte, E.M. A Bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS Biol. 2013, 11, e1001624. [Google Scholar] [CrossRef]
- Hamanaka, K.; Takata, A.; Uchiyama, Y.; Miyatake, S.; Miyake, N.; Mitsuhashi, S.; Iwama, K.; Fujita, A.; Imagawa, E.; Alkanaq, A.N.; et al. MYRF haploinsufficiency causes 46,XY and 46,XX disorders of sex development: Bioinformatics consideration. Hum. Mol. Genet. 2019, 28, 2319–2329. [Google Scholar] [CrossRef]
- Qi, H.; Yu, L.; Zhou, X.; Wynn, J.; Zhao, H.; Guo, Y.; Zhu, N.; Kitaygorodsky, A.; Hernan, R.; Aspelund, G.; et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018, 14, e1007822. [Google Scholar] [CrossRef]
- Pinz, H.; Pyle, L.C.; Li, D.; Izumi, K.; Skraban, C.; Tarpinian, J.; Braddock, S.R.; Telegrafi, A.; Monaghan, K.G.; Zackai, E.; et al. De novo variants in Myelin regulatory factor (MYRF) as candidates of a new syndrome of cardiac and urogenital anomalies. Am. J. Med. Genet. A 2018, 176, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, H.; Azuma, Y.; Masuda, A.; Okuno, T.; Nakahara, E.; Imamura, T.; Saitoh, M.; Mizuguchi, M.; Shimizu, T.; Ohno, K.; et al. MYRF is associated with encephalopathy with reversible myelin vacuolization. Ann. Neurol. 2018, 83, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Garnai, S.J.; Brinkmeier, M.L.; Emery, B.; Aleman, T.S.; Pyle, L.C.; Veleva-Rotse, B.; Sisk, R.A.; Rozsa, F.W.; Ozel, A.B.; Li, J.Z.; et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. 2019, 15, e1008130. [Google Scholar] [CrossRef]
- Wilczewski, C.M.; Obasohan, J.; Paschall, J.E.; Zhang, S.; Singh, S.; Maxwell, G.L.; Similuk, M.; Wolfsberg, T.G.; Turner, C.; Biesecker, L.G.; et al. Genotype first: Clinical genomics research through a reverse phenotyping approach. Am. J. Hum. Genet. 2023, 110, 3–12. [Google Scholar] [CrossRef]
- Kaplan, J.D.; Stewart, B.; Prasov, L.; Pyle, L.C. MYRF-Related Cardiac Urogenital Syndrome. In GeneReviews; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Rossetti, L.Z.; Glinton, K.; Yuan, B.; Liu, P.; Pillai, N.; Mizerik, E.; Magoulas, P.; Rosenfeld, J.A.; Karaviti, L.; Sutton, V.R.; et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genet. A 2019, 179, 1376–1382. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correa Brito, L.; Grinspon, R.P.; Lopez Dacal, J.; Scaglia, P.; Esnaola Azcoiti, M.; Izquierdo, A.; Ropelato, M.G.; Rey, R.A. Identification of a Novel Variant in Myelin Regulatory Growth Factor by Next-Generation Sequencing Led to the Detection of a Clinically Inapparent Congenital Heart Defect in a Patient with a 46,XY Disorder of Sex Development. J. Pers. Med. 2023, 13, 1158. https://doi.org/10.3390/jpm13071158
Correa Brito L, Grinspon RP, Lopez Dacal J, Scaglia P, Esnaola Azcoiti M, Izquierdo A, Ropelato MG, Rey RA. Identification of a Novel Variant in Myelin Regulatory Growth Factor by Next-Generation Sequencing Led to the Detection of a Clinically Inapparent Congenital Heart Defect in a Patient with a 46,XY Disorder of Sex Development. Journal of Personalized Medicine. 2023; 13(7):1158. https://doi.org/10.3390/jpm13071158
Chicago/Turabian StyleCorrea Brito, Lourdes, Romina P. Grinspon, Jimena Lopez Dacal, Paula Scaglia, María Esnaola Azcoiti, Agustín Izquierdo, María Gabriela Ropelato, and Rodolfo A. Rey. 2023. "Identification of a Novel Variant in Myelin Regulatory Growth Factor by Next-Generation Sequencing Led to the Detection of a Clinically Inapparent Congenital Heart Defect in a Patient with a 46,XY Disorder of Sex Development" Journal of Personalized Medicine 13, no. 7: 1158. https://doi.org/10.3390/jpm13071158
APA StyleCorrea Brito, L., Grinspon, R. P., Lopez Dacal, J., Scaglia, P., Esnaola Azcoiti, M., Izquierdo, A., Ropelato, M. G., & Rey, R. A. (2023). Identification of a Novel Variant in Myelin Regulatory Growth Factor by Next-Generation Sequencing Led to the Detection of a Clinically Inapparent Congenital Heart Defect in a Patient with a 46,XY Disorder of Sex Development. Journal of Personalized Medicine, 13(7), 1158. https://doi.org/10.3390/jpm13071158