
The Value of Multimodal Imaging in Early Phenotyping of Cardiomyopathies: A Family Case Report

 , ,
, ,
Abstract
1. Introduction
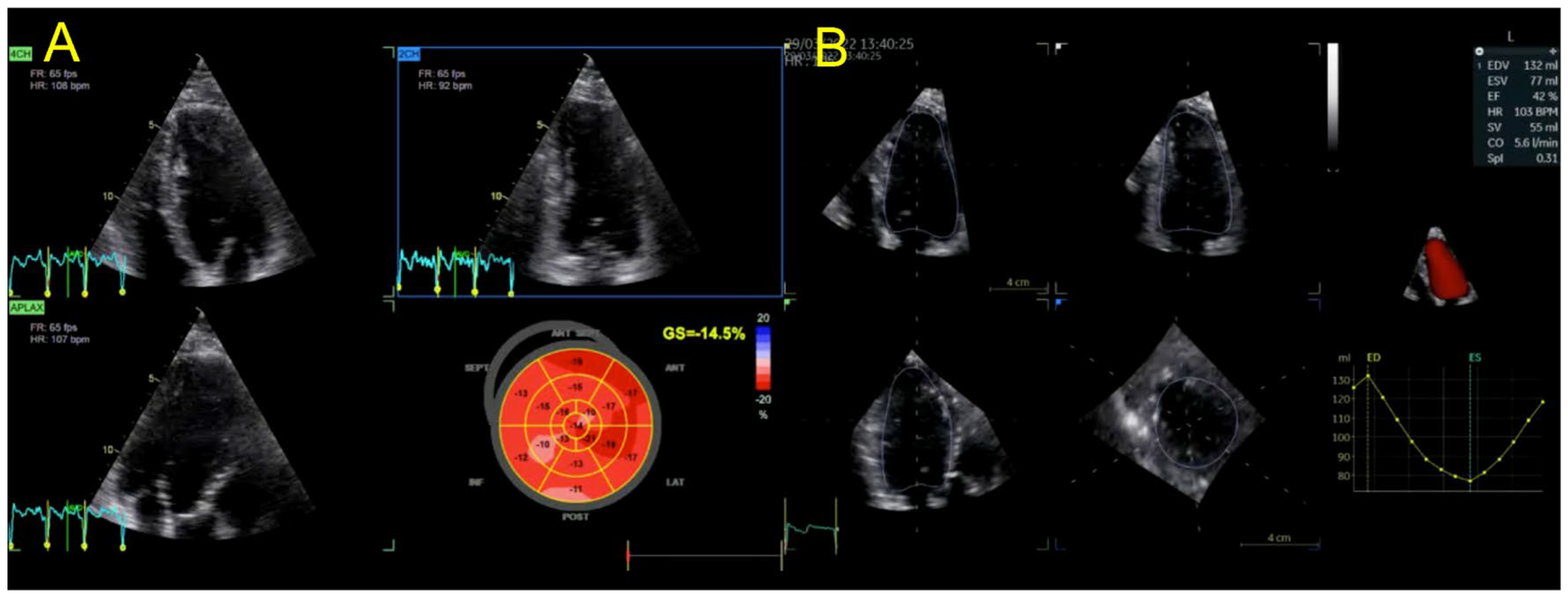
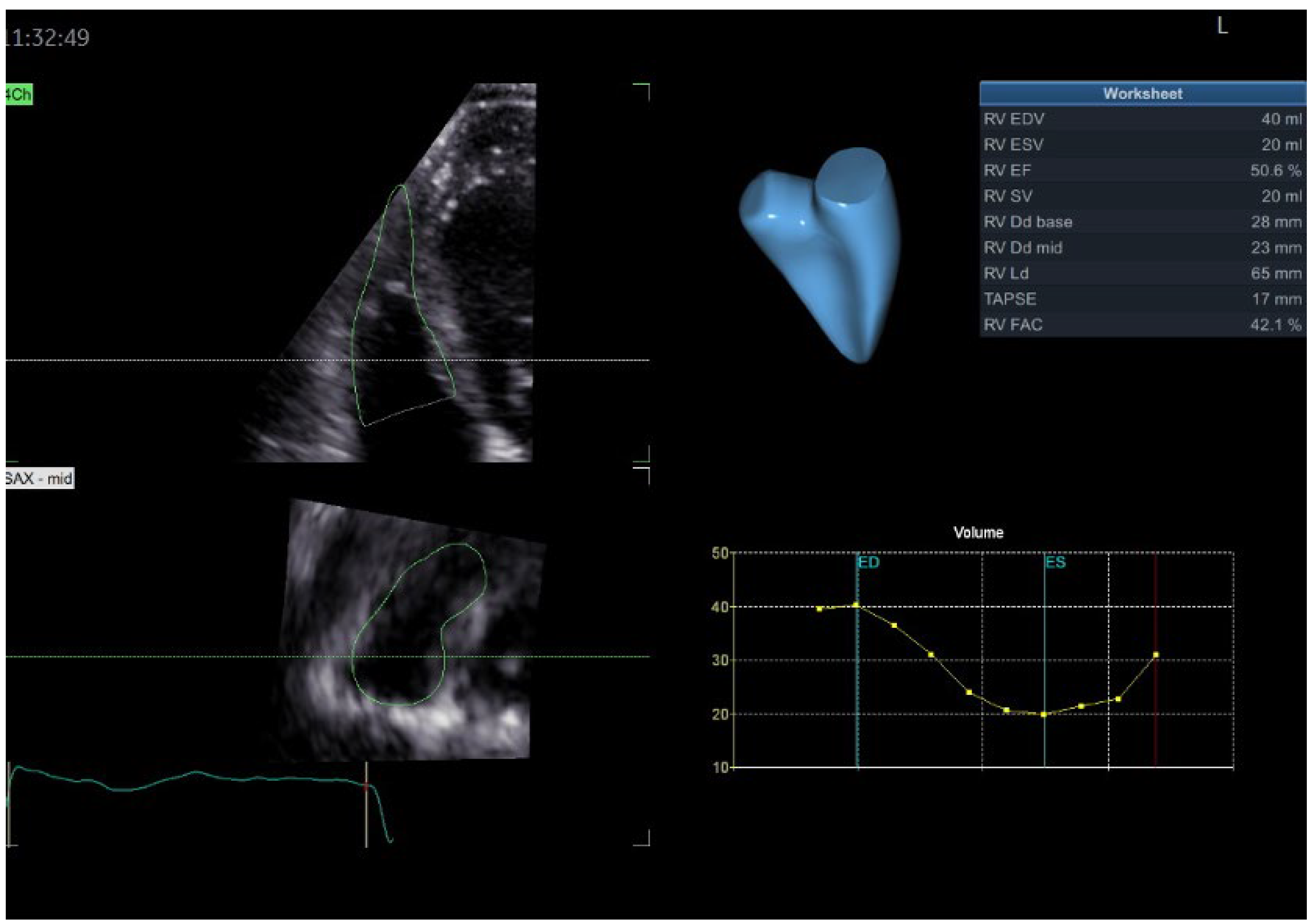
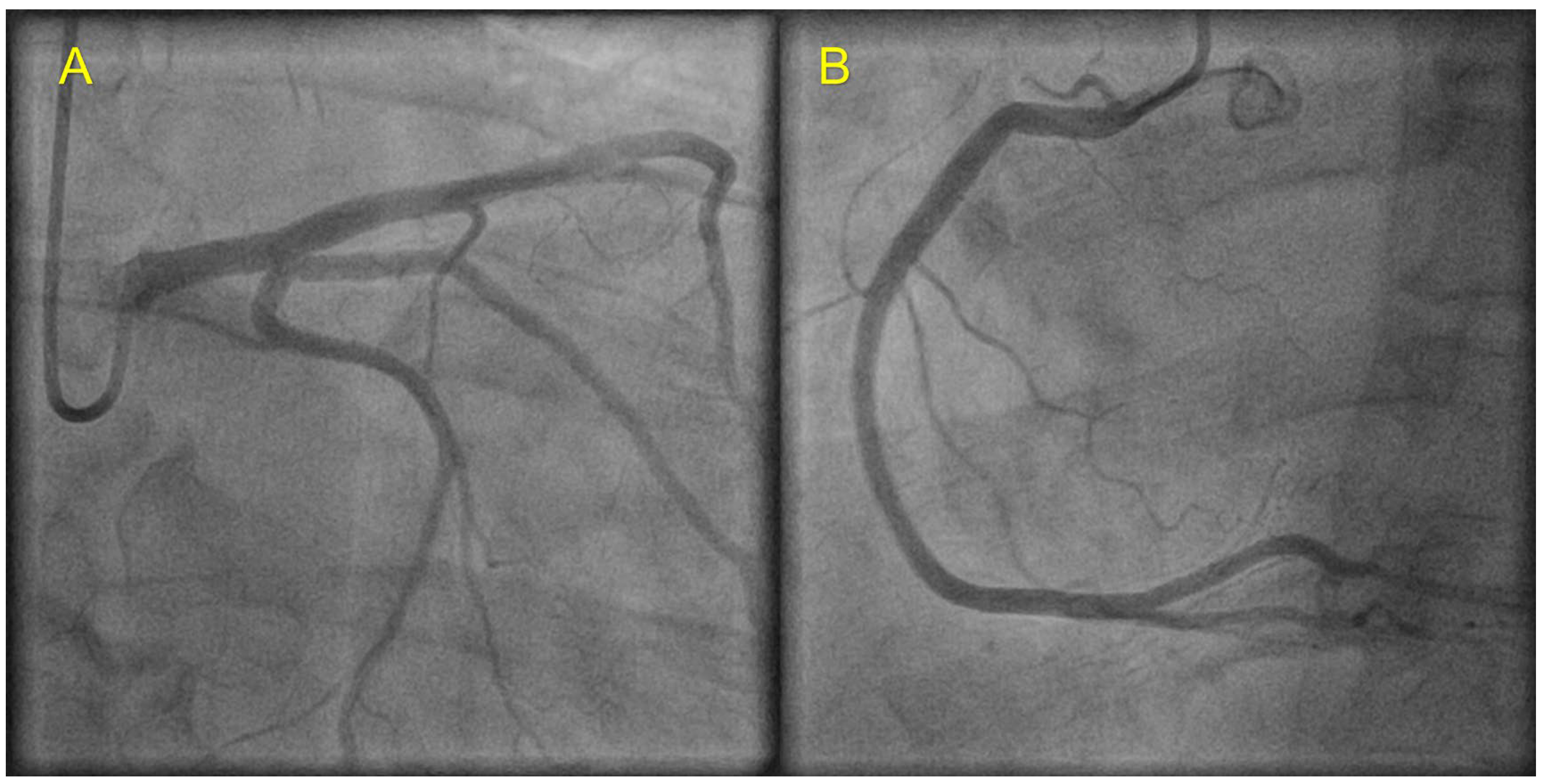
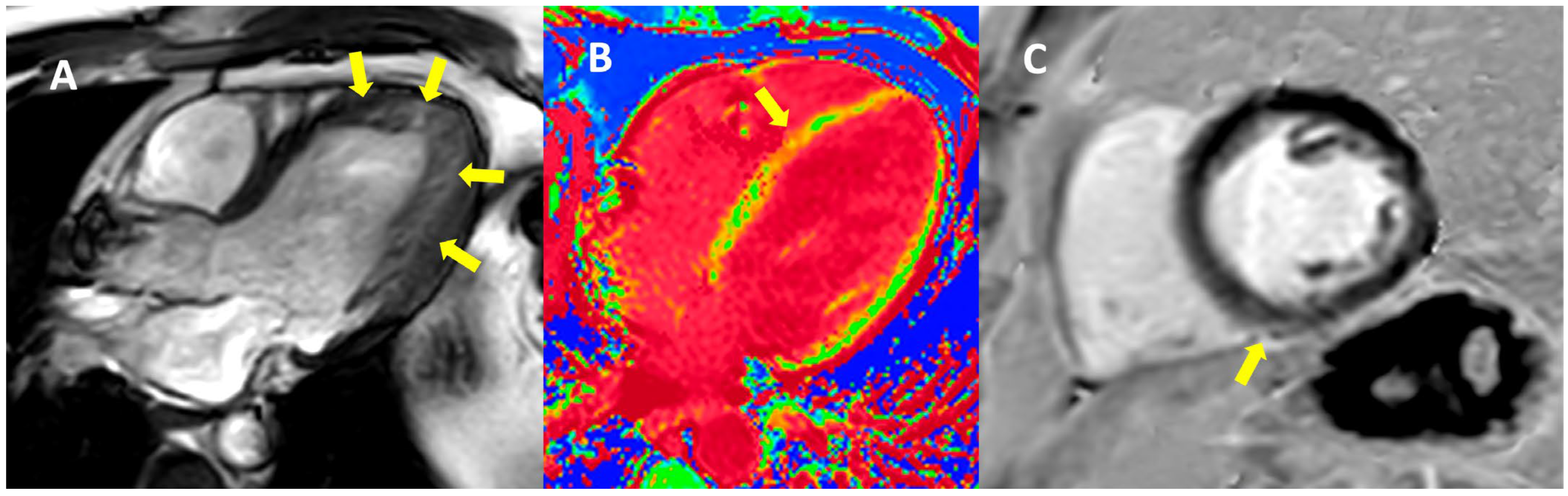
2. Case Presentation
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a Revised Definition of Dilated Cardiomyopathy, Hypokinetic Non-Dilated Cardiomyopathy, and Its Implications for Clinical Practice: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving Concepts in Dilated Cardiomyopathy. Eur. J. Heart Fail 2018, 20, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, Z.; Guo, L.; Yu, S.; Li, T.; Zheng, L.; Pan, G.; Yang, J.; Sun, Y.; Hui, R.; et al. Prevalence of Hypokinetic Non-Dilated Cardiomyopathy in a Large General Chinese Population. Int. J. Cardiol. 2016, 223, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Yukimitsu, N.; Yokoyama, N.; Ikeda, Y.; Ishibashi, R.; Takamura, S.; Kozuma, K.; Hatsuno, M. One-year outcome in hypokinetic non-dilated cardiomyopathy detected by cardiac magnetic resonance: Idiopathic dilated cardiomyopathy comparison. J. Am. Coll. Cardiol. 2022, 79, 359. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Merlo, M.; Barbati, G.; Ramani, F.; Brun, F.; Pinamonti, B.; Sinagra, G. Insights into Mildly Dilated Cardiomyopathy: Temporal Evolution and Long-Term Prognosis. Eur. J. Heart Fail 2017, 19, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Graziosi, M.; Ditaranto, R.; Rapezzi, C.; Pasquale, F.; Lovato, L.; Leone, O.; Parisi, V.; Potena, L.; Ferrara, V.; Minnucci, M.; et al. Clinical Presentations Leading to Arrhythmogenic Left Ventricular Cardiomyopathy. Open Heart 2022, 9, e001914. [Google Scholar] [CrossRef] [PubMed]
- Casas, G.; Limeres, J.; Oristrell, G.; Gutierrez-Garcia, L.; Andreini, D.; Borregan, M.; Larrañaga-Moreira, J.M.; Lopez-Sainz, A.; Codina-Solà, M.; Teixido-Tura, G.; et al. Clinical Risk Prediction in Patients with Left Ventricular Myocardial Noncompaction. J. Am. Coll. Cardiol. 2021, 78, 643–662. [Google Scholar] [CrossRef] [PubMed]
- Mirea, O.; Berceanu, M.; Constantin, A.; Mănescu, M.; Târtea, G.C.; Donoiu, I.; Militaru, C.; Istrătoaie, O. Non-compaction cardiomyopathy–brief review. J. Mind Med. Sci. 2017, 4, 115–124. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) Variants in Chemotherapy-Induced Cardiomyopathy. J. Card Fail 2017, 23, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, E.J.; Takayama, H.; Ferrari, G. Stretch Your Heart—But Not Too Far: The Role of Titin Mutations in Dilated Cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2018, 156, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, E.N.; Jenni, R. Left ventricular non-compaction revisited: A distinct phenotype with genetic heterogeneity? Eur. Heart J. 2011, 32, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Paterick, T.E.; Tajik, A.J. Left Ventricular Noncompaction—A Diagnostically Challenging Cardiomyopathy. Circ. J. 2012, 76, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Halliday, B.P.; Baksi, A.J.; Gulati, A.; Ali, A.; Newsome, S.; Izgi, C.; Arzanauskaite, M.; Lota, A.; Tayal, U.; Vassiliou, V.S.; et al. Outcome in Dilated Cardiomyopathy Related to the Extent, Location, and Pattern of Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2019, 12, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Walsh, R.; Barton, P.J.; Ware, J.S.; Cook, S.A.; Prasad, S.K. Truncating Variants in Titin Independently Predict Early Arrhythmias in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 69, 2466–2468. [Google Scholar] [CrossRef] [PubMed]
- Corden, B.; Jarman, J.; Whiffin, N.; Tayal, U.; Buchan, R.; Sehmi, J.; Harper, A.; Midwinter, W.; Lascelles, K.; Markides, V.; et al. Association of Titin-Truncating Genetic Variants with Life-threatening Cardiac Arrhythmias in Patients with Dilated Cardiomyopathy and Implanted Defibrillators. JAMA Netw. Open 2019, 6, e196520. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proband | Brother | Nephew | |
|---|---|---|---|
| Clinical | Arterial hypertension Dyslipidemia | NYHA II HFrEF Persistent atrial fibrillation Arterial hypertension | Anabolic steroid use Resistance training Arterial hypertension |
| Echocardiography | Hypokinetic non-dilated cardiomyopathy LVEF = 42% Mild mitral regurgitation IAS aneurysm | Hypokinetic non-dilated cardiomyopathy LVEF = 34% Mild mitral regurgitation IAS aneurysm | Left ventricular concentric hypertrophy LVEF = 54% |
| Cardiac magnetic resonance imaging | Hypokinetic non-dilated cardiomyopathy Left ventricular non-compaction LVEF = 50% | Hypokinetic non-dilated cardiomyopathy Regional sub-epicardial fibrosis LVEF = 41% | Not carried out |
| Coronary angiography | Normal | Normal | Not carried out |
| Genetic testing | Heterozygous TTN variant—deletion in exon 3 (exon 326, c.70482_70483del p.Tyr23494*) | Heterozygous TTN variant—deletion in exon 3 (exon 326, c.70482_70483del p.Tyr23494*) | No pathogenic/likely pathogenic mutations identified |
| Management | CV risk factor control HTN treatment optimization Clinical and echo follow up | CV risk factor control HFrEF treatment optimization Clinical and echo follow up after 3 months of OMT | CV risk factor control Cessation of steroid use Clinical and echo follow up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iovănescu, M.L.; Hădăreanu, D.R.; Militaru, S.; Florescu, C.; Militaru, C.; Donoiu, I. The Value of Multimodal Imaging in Early Phenotyping of Cardiomyopathies: A Family Case Report. J. Pers. Med. 2023, 13, 742. https://doi.org/10.3390/jpm13050742
Iovănescu ML, Hădăreanu DR, Militaru S, Florescu C, Militaru C, Donoiu I. The Value of Multimodal Imaging in Early Phenotyping of Cardiomyopathies: A Family Case Report. Journal of Personalized Medicine. 2023; 13(5):742. https://doi.org/10.3390/jpm13050742
Chicago/Turabian StyleIovănescu, Maria Livia, Diana Ruxandra Hădăreanu, Sebastian Militaru, Cristina Florescu, Constantin Militaru, and Ionuț Donoiu. 2023. "The Value of Multimodal Imaging in Early Phenotyping of Cardiomyopathies: A Family Case Report" Journal of Personalized Medicine 13, no. 5: 742. https://doi.org/10.3390/jpm13050742
APA StyleIovănescu, M. L., Hădăreanu, D. R., Militaru, S., Florescu, C., Militaru, C., & Donoiu, I. (2023). The Value of Multimodal Imaging in Early Phenotyping of Cardiomyopathies: A Family Case Report. Journal of Personalized Medicine, 13(5), 742. https://doi.org/10.3390/jpm13050742