
Image Analysis in Digital Pathology Utilizing Machine Learning and Deep Neural Networks †



Abstract
:1. Introduction
2. Related Work and Background Information
2.1. The Concept of Digital Pathology (DP)
2.2. State-of-the-Art Computer-Based Methodologies and Tools for Digital Pathology
2.2.1. Computer Vision
2.2.2. Machine Learning
2.2.3. Feature Extraction and Selection
2.2.4. Deep Learning in Computer Vision
2.2.5. Existing Approaches vs. Deep Learning
3. Material and Methods
3.1. Methodology and Implementation
Deep Learning Framework Specifications
3.2. Use Cases Description
3.2.1. Object Detection on TUPAC16
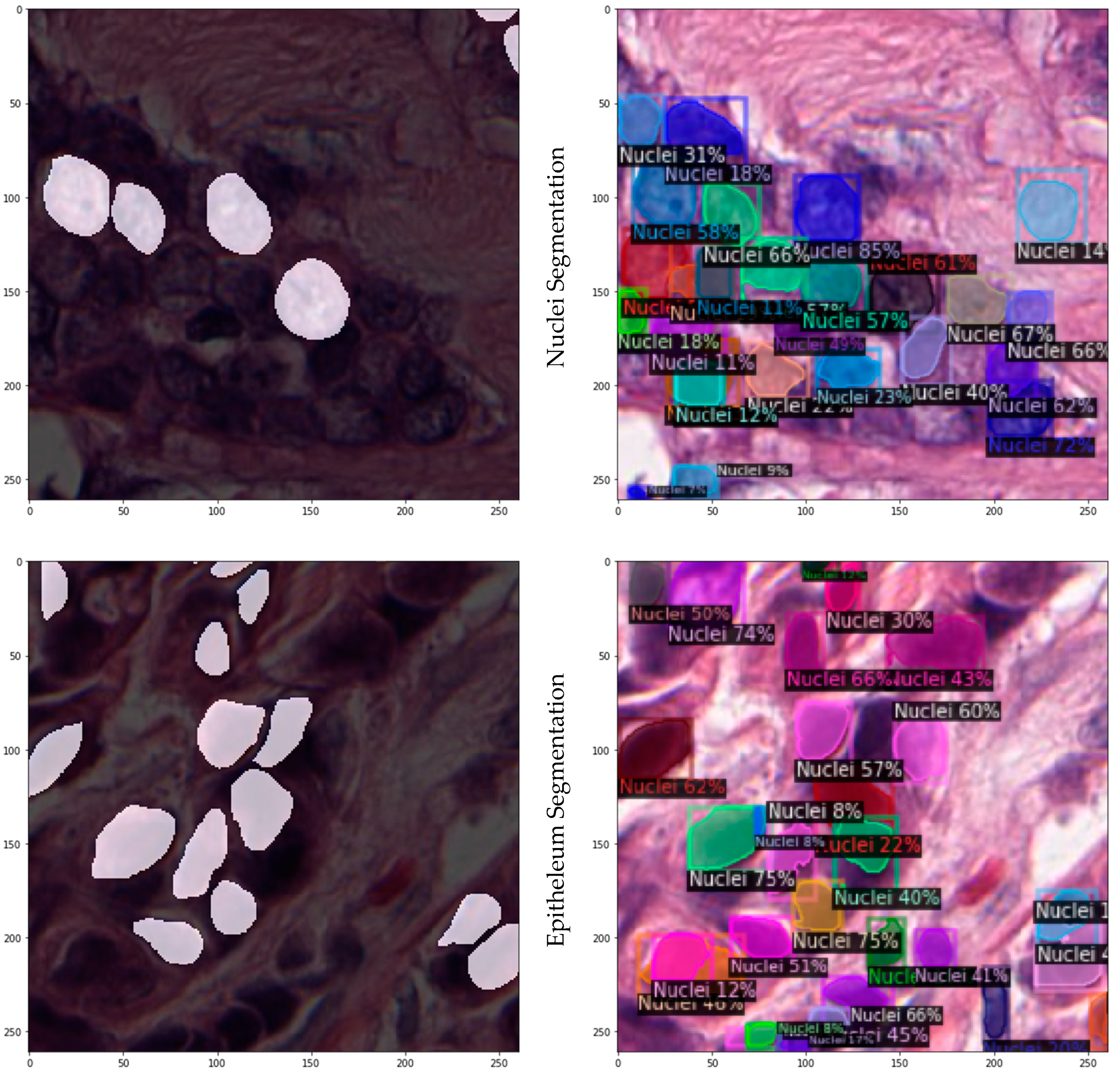
3.2.2. Instance Segmentation on JPATHOL
3.2.3. Data Processing and Consumption
3.2.4. Parameterization and Training
4. Experimental Results
4.1. TUPAC16
4.2. JPATHOL
4.3. Comparison with SOTA
5. Discussion and Conclusions
5.1. Future Work
5.2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Madabhushi, A. Digital pathology image analysis: Opportunities and challenges. Imaging Med. 2009, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Veta, M.; Heng, Y.J.; Stathonikos, N.; Bejnordi, B.E.; Beca, F.; Wollmann, T.; Rohr, K.; Shah, M.A.; Wang, D.; Rousson, M.; et al. Predicting breast tumor proliferation from whole-slide images: The TUPAC16 challenge. Med. Image Anal. 2019, 54, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ballard, D.H. Computer Vision; Prentice Hall: Hoboken, NJ, USA, 1982; ISBN 978-0-13-165316-0. [Google Scholar]
- Huang, T. Computer Vision: Evolution and Promise. In Proceedings of the 19th CERN School of Computing, Egmond aan Zee, The Netherlands, 8–21 September 1996; CERN: Geneva, Switzerland, 1996; pp. 21–25, ISBN 978-9290830955. [Google Scholar]
- Goudas, T.; Maglogiannis, I. An Advanced Image Analysis Tool for the Quantification and Characterization of Breast Cancer in Microscopy Images. J. Med. Syst. 2015, 39, 13. Available online: https://www.scopus.com/inward/record.uri?eid=2-s2.0-84923112995&doi=10.1007%2fs10916-015-0225-3&partnerID=40&md5=959c05a8d8e08777d21c3531fe3470f4 (accessed on 25 August 2022). [CrossRef] [PubMed]
- Sonka, M.; Hlavac, V.; Boyle, R. Image Processing, Analysis, and Machine Vision; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Erickson, B.J. Machine learning for medical imaging. Radiographics 2017, 37, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Chaochao, L.; Tang, X. Surpassing Human-Level Face Verification Performance on LFW with GaussianFace. In Proceedings of the 29th AAAI Conference on Artificial Intelligence (AAAI-15), Austin, TX, USA, 25–30 January 2015. [Google Scholar]
- Anagnostopoulos, I.; Maglogiannis, I. Neural network-based diagnostic and prognostic estimations in breast cancer microscopic instances. Med. Biol. Eng. Comput. 2006, 44, 773–784. Available online: https://www.scopus.com/inward/record.uri?eid=2-s2.0-33748514800&doi=.org/10.1007%2f/s11517-006-0079-4&partnerID=40&md5=34089a4b6f94144b8e1a143e50593ec6 (accessed on 25 August 2022). [CrossRef] [PubMed]
- Maglogiannis, I.; Sarimveis, H.; Kiranoudis, C.T.; Chatziioannou, A.A.; Oikonomou, N.; Aidinis, V. Radial Basis Function Neural Networks Classification for the Recognition of Idiopathic Pulmonary Fibrosis in Microscopic Images. IEEE Trans. Inf. Technol. Biomed. 2008, 12, 42–54. Available online: https://www.scopus.com/inward/record.uri?eid=2-s2.0-39449139992&doi=10.1109%2fTITB.2006.888702&partnerID=40&md5=92e5362c8bab3acd12cd1080fd882e64 (accessed on 25 August 2022). [CrossRef] [PubMed]
- Kallipolitis, A.; Stratigos, A.; Zarras, A.; Maglogiannis, I. Fully connected visual words for the classification of skin cancer confocal images. In Proceedings of the VISIGRAPP 2020—15th International Joint Conference on Computer Vision, Imaging and Computer Graphics Theory and Applications, Valletta, Malta, 27–29 February 2020; Volume 5, pp. 853–858. Available online: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85083509307&partnerID=40&md5=3a0b72b8923469044bd671eeb51596e3 (accessed on 25 August 2022).
- Maglogiannis, I.; Delibasis, K.K. Enhancing classification accuracy utilizing globules and dots features in digital dermoscopy. Comput. Methods Programs Biomed. 2015, 118, 124–133. Available online: https://www.scopus.com/inward/record.uri?eid=2-s2.0-84922445570&doi=10.1016%2fj.cmpb.2014.12.001&partnerID=40&md5=4efa18dd5204e726d4a3d468b4ac933d (accessed on 25 August 2022). [CrossRef] [PubMed]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- O’ Mahony, N.; Campbell, S.; Carvalho, A.; Harapanahalli, S.; Hernandez, G.V.; Krpalkova, L.; Riordan, D.; Walsh, J. Deep Learning vs. Traditional Computer Vision. In Proceedings of the 2019 Computer Vision Conference (CVC)—Advances in Computer Vision, Las Vegas, NV, USA, 25–26 April 2019. [Google Scholar]
- O’Mahony, N.C. Deep learning vs. traditional computer vision. In Science and Information Conference; Springer: Cham, Switzerland, 2019; pp. 128–144. [Google Scholar]
- Kayalibay, B.; Jensen, G.; van der Smagt, P. CNN-based segmentation of medical imaging data. arXiv 2017, arXiv:1701.03056. [Google Scholar]
- Kraus, O.Z.; Ba, J.L.; Frey, B.J. Classifying and segmenting microscopy images with deep multiple instance learning. Bioinformatics 2016, 32, i52–i59. [Google Scholar] [CrossRef] [PubMed]
- Janowczyk, A.; Madabhushi, A. Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases. J. Pathol. Inform. 2016, 7, 29. [Google Scholar] [CrossRef]
- Bay, H.; Tuytelaars, T.; Gool, L.V. SURF: Speeded Up Robust Features. In Computer Vision and Image Understanding (CVIU), Volume 110, No. 3; Springer: Berlin/Heidelberg, Germany, 2008; pp. 346–359. [Google Scholar]
- Lowe, D.G. Distinctive Image Features from Scale-Invariant Keypoints. Int. J. Comput. Vis. 2004, 60, 91–110. [Google Scholar] [CrossRef]
- Wu, Y.; Kirillov, A.; Massa, F.; Lo, W.Y.; Girshick, R. Detectron2. 2019. Available online: https://github.com/facebookresearch/detectron2 (accessed on 25 August 2022).
- Lin, T.Y.; Maire, M.; Belongie, S.; Bourdev, L.; Girshick, R.; Hays, J.; Perona, P.; Zitnick, C.L.; Dollár, P. Microsoft COCO: Common Objects in Context. arXiv 2015, arXiv:1405.0312. [Google Scholar]
- He, K.; Gkioxari, G.; Dollár, P.; Girshick, R. Mask R-CNN. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 2961–2969. [Google Scholar]
- Ren, S.; He, K.; Girshick, R.; Sun, J. Faster R-CNN: Towards Real-Time Object Detection with Region Proposal Networks. Adv. Neural Inf. Process. Syst. 2016, 39, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Van Diest, P.J. Prognostic value of proliferation in invasive breast cancer: A review. J. Clin. Pathol. 2004, 57, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, P.L.; Page, D.L.; Weaver, D.; Thor, A.D.; Allred, D.C.; Clark, G.M.; Ruby, S.G.; O’Malley, F.; Simpson, J.F.; Connolly, J.L.; et al. Prognostic factors in breast cancer. College of American Pathologists Consensus Statement 1999. Arch. Pathol. Lab. Med. 2000, 124, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Roux, L. MITOS-ATYPIA-14 Contest Home Page. 2014. Available online: https://mitos-atypia-14.grand-challenge.org/ (accessed on 25 August 2022).
- Veta, M.; van Diest, P.J.; Willems, S.M.; Wang, H.; Madabhushi, A.; Cruz-Roa, A.; Gonzalez, F.; Larsen, A.B.; Vestergaard, J.S.; Dahl, A.B.; et al. Assessment of algorithms for mitosis detection in breast cancer histopathology images. Med. Image Anal. 2015, 20, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.H.; Sangoi, A.R.; Leung, S.; Marinelli, R.J.; Nielsen, T.O.; van de Vijver, M.J.; West, R.B.; van de Rijn, M.; Koller, D. Systematic Analysis of Breast Cancer Morphology Uncovers Stromal Features Associated with Survival. Sci. Transl. Med. 2011, 3, 108ra113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macenko, M.; Niethammer, M.; Marron, J.S.; Borland, D.; Woosley, J.T.; Guan, X.; Schmitt, C.; Thomas, N.E. A method for normalizing histology slides for quantitative analysis. In Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Boston, MA, USA, 28 June–1 July 2009; pp. 1107–1110. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- IMAG/e, M.I.; Tumor Proliferation Assessment Challenge 2016. Results Task 3: Mitosis Detection. 2016. Available online: http://tupac.tue-image.nl/node/62 (accessed on 25 May 2021).
- Cireşan, D.C. Mitosis detection in breast cancer histology images with deep neural networks. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Nagoya, Japan, 22–26 September 2013; Springer: Berlin, Germany, 2013; pp. 411–418. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Biological Motivation | Dataset |
|---|---|---|
| Nuclei segmentation | Pleomorphism is used in current clinical grading schemes | 141 × 2000 × 2000 @40× ROIs of ER+ BCa, containing subset of 12,000 annotated nuclei |
| Epithelium segmentation | Epithelium regions contribute to identification of tumor infiltrating lymphocytes (TILs) | 42 × 1000 × 1000 @20× ROIs from ER+ BCa, containing 1735 regions |
| Tubule segmentation | Area estimates in high power fields are critical in BCa grading schemes | 85 × 775 × 522 @40× ROIs from colorectal cancer, containing 795 delineated tubules |
| Layer | Type | Kernels | Kernel Size | Stride | Activation |
|---|---|---|---|---|---|
| 0 | Input | 3 | 32 × 32 | ||
| 1 | Convolution | 32 | 5 × 5 | 1 | |
| 2 | Max pool | 3 × 3 | 2 | ReLU | |
| 3 | Convolution | 32 | 5 × 5 | 1 | ReLU |
| 4 | Mean pool | 3 × 3 | 2 | ||
| 5 | Convolution | 64 | 5 × 5 | 1 | ReLU |
| 6 | Mean pool | 3 × 3 | 2 | ||
| 7 | Fully connected | 64 | Dropout + ReLU | ||
| 8 | Fully connected | 2 | Dropout + ReLU | ||
| 9 | SoftMax |
| Variable | Setting |
|---|---|
| Batch size | 128 |
| Learning rate | 0.001 |
| Learning rate schedule | Adagrad |
| Rotations | 0, 90 |
| Num. iterations | 600,000 |
| Weight decay | 0.004 |
| Random minor | Enabled |
| Transformation | Mean-centered |
| Hyperparameter | Tuning Range | Starting Value | Optimum Value |
|---|---|---|---|
| Number of workers | (2, 8) | 2 | 8 |
| Images per batch | (2, 8) | 2 | 4 |
| Learning rate | (0.00025, 0.1) | 0.00025 | 0.025 |
| Max iterations | (300, 12,000) | 300 | 9000 |
| Batch size per image | (128, 1024]) | 128 | 512 |
| Testing threshold | (0.01, 0.7) | 0.7 | 0.07 |
| Bounding box dimension | (40, 120) | 80 | 80 |
| Subimage split dimension | (224, 1000) | 1000 | 500 |
| Images/ Batch | Learning Rate | Max Iterations | RoIHead Batch Size | Split Dimension | Train Time (H:M:S) | AP (%) | STDEV (±%) |
|---|---|---|---|---|---|---|---|
| 4 | 0.025 | 600 | 512 | 1000 × 1000 | 0:04:56 | 43.32 | 6.58 |
| 4 | 0.025 | 600 | 1024 | 1000 × 1000 | 0:05:23 | 40.11 | 8.65 |
| 8 | 0.025 | 600 | 1024 | 1000 × 1000 | 0:11:35 | 43.79 | 5.69 |
| 6 | 0.010 | 9000 | 1024 | 1000 × 1000 | 2:02:55 | 55.74 | 3.26 |
| 4 | 0.025 | 9000 | 512 | 1000 × 1000 | 1:13:18 | 58.11 | 2.96 |
| 4 | 0.025 | 9000 | 512 | 500 × 500 | 1:11:49 | 65.02 | 4.05 |
| Dataset | AP (%) | STDEV (±%) |
|---|---|---|
| Nuclei | 19.53 | 1.68 |
| Epithelium | 5.15 | 3.21 |
| Tubule | 35.22 | 5.16 |
| Nuc/Epi/Tub | 8.03 | 2.09 |
| AP | AP50 | AP75 | Aps | Apm | Apl | AP-Nuc | AP-Epi | AP-Tub |
|---|---|---|---|---|---|---|---|---|
| 14.41 | 23.72 | 15.31 | 5.16 | 22.42 | 17.67 | 15.26 | 2.06 | 25.91 |
| Method | Detection | F-Score |
|---|---|---|
| 20× | 0.95 | 0.80 |
| 20× + dropout | 0.90 | 0.79 |
| 40× | 0.98 | 0.83 |
| Baseline model—40× | 0.14 | 0.220 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amerikanos, P.; Maglogiannis, I. Image Analysis in Digital Pathology Utilizing Machine Learning and Deep Neural Networks. J. Pers. Med. 2022, 12, 1444. https://doi.org/10.3390/jpm12091444
Amerikanos P, Maglogiannis I. Image Analysis in Digital Pathology Utilizing Machine Learning and Deep Neural Networks. Journal of Personalized Medicine. 2022; 12(9):1444. https://doi.org/10.3390/jpm12091444
Chicago/Turabian StyleAmerikanos, Paris, and Ilias Maglogiannis. 2022. "Image Analysis in Digital Pathology Utilizing Machine Learning and Deep Neural Networks" Journal of Personalized Medicine 12, no. 9: 1444. https://doi.org/10.3390/jpm12091444
APA StyleAmerikanos, P., & Maglogiannis, I. (2022). Image Analysis in Digital Pathology Utilizing Machine Learning and Deep Neural Networks. Journal of Personalized Medicine, 12(9), 1444. https://doi.org/10.3390/jpm12091444