Abstract
Polygenic models have emerged as promising prediction tools for the prediction of complex traits. Currently, the majority of polygenic models are developed in the context of predicting disease risk, but polygenic models may also prove useful in predicting drug outcomes. This study sought to understand how polygenic models incorporating pharmacogenetic variants are being used in the prediction of drug outcomes. A systematic review was conducted with the aim of gaining insights into the methods used to construct polygenic models, as well as their performance in drug outcome prediction. The search uncovered 89 papers that incorporated pharmacogenetic variants in the development of polygenic models. It was found that the most common polygenic models were constructed for drug dosing predictions in anticoagulant therapies (n = 27). While nearly all studies found a significant association with their polygenic model and the investigated drug outcome (93.3%), less than half (47.2%) compared the performance of the polygenic model against clinical predictors, and even fewer (40.4%) sought to validate model predictions in an independent cohort. Additionally, the heterogeneity of reported performance measures makes the comparison of models across studies challenging. These findings highlight key considerations for future work in developing polygenic models in pharmacogenomic research.
1. Introduction
The concept of polygenic inheritance was first introduced in 1918 by R.A. Fisher who showed that continuous traits are passed down through Mendelian inheritance of many genetic variants of small effect [1]. Since then, this polygenic approach to inheritance has been used to study complex human phenotypes [2,3,4,5,6]. Given the small individual effects that each genetic variant contributes to the heritability of complex traits, polygenic scores have emerged as tools to estimate individual probability for these complex phenotypes. Polygenic scores combine the individual effects of several genetic variants into a single score which can be used to assign a probability to any individual representing their genetic predisposition for a phenotype [7,8,9]. As genotyping technologies become increasingly affordable, the excitement surrounding the possibility of generating genome-wide risk scores for various diseases is continually growing [7,10].
Thus far, polygenic scores have primarily been applied in the prediction of disease risk. A highly cited study by Khera et al., published in 2018, developed a polygenic risk score comprised of over 6 million common single nucleotide polymorphisms (SNPs) to predict individual risk of developing coronary artery disease (CAD) [9]. The higher the burden of risk-alleles, the higher an individual’s genetic risk of CAD, and one of the striking discoveries in this study was that patients within the top 8% of the polygenic risk score had a 3-fold increased risk of CAD which is comparable to the risk imparted by rare, monogenic causes of heart disease [9]. The advantage of the polygenic approach is that because it is constructed using common SNPs, it can be applied to many more patients, whereas only a small proportion of the population will carry rare genetic variants. Many other similar polygenic scores have been developed to predict disease risk, and thus, polygenic scores offer the potential to improve genetic screening for disease and are more generalizable to the broader population [11,12].
The polygenic nature of complex traits and disease have become widely accepted, but this has not been translated to the same extent within the field of pharmacogenomics [13]. Innumerable genetic studies have been conducted to explain the interindividual variability in drug-related outcomes such as nonresponse, dosing requirements, and the development of adverse drug reactions (ADRs) [13]. However, many of these early pharmacogenetic studies focused on the monogenic architecture of drug-outcomes where genetic variants of larger effect size were thought of as separate predictors [13]. Relatively few studies have aimed to combine pharmacogenetic variants to improve predictions of these drug outcomes. Perhaps one of the most well-studied multigenic-drug interactions is in warfarin dosing. Warfarin is a widely used oral anticoagulant with a narrow therapeutic window and a high interindividual variability in dosing requirements [14,15]. Early genome-wide association studies of warfarin maintenance dose identified pharmacogenetic variants in VKORC1 and CYP2C9 which were strongly associated with warfarin dosing requirements, and genotyping for these variants have been added to the FDA warfarin dosing guidelines [16,17,18]. This highlights the potential utility of pharmacogenetic prediction models comprising multiple genetic loci to guide treatment decisions in clinical practice.
Polygenic scores in pharmacogenomics research were recently reviewed, examining the use of polygenic scores developed from pre-existing genetic studies in disease phenotypes as a predictor of drug outcomes (e.g., schizophrenia-derived polygenic risk score used to predict lurasidone response) [19,20]. However, there has been no review to-date evaluating the use of polygenic models derived specifically from pharmacogenetic variants associated with gene-drug relationships. To this end, a systematic review was conducted to summarize the methods and performance of polygenic models encompassing pharmacogenetic variants in predicting drug outcome phenotypes. In the context of this review, a polygenic model was broadly defined as any model or score encompassing pharmacogenetic variants at more than one genetic locus. In doing so, this review aims to understand the current methods used to develop polygenic models for predicting drug outcomes, as well as the performance of these models in their ability to reliably predict drug outcomes in patients.
2. Materials and Methods
The Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines for a systematic review were followed to ensure completeness of the review. A study protocol was written prior to the initiation of the review but was not registered.
2.1. Rationale and Scope of Review
This review aimed to summarize the methods and performance of polygenic models encompassing pharmacogenetic variants for predicting drug outcomes. While polygenic models encompassing non-pharmacogenetic variants have been applied to the prediction of drug outcomes, these models were not considered to be true pharmacogenetic models and were not included in the current review. An example of one such article is a 2018 study by Li et al., examining the association between a polygenic risk score derived from schizophrenia risk-alleles and response to lurasidone treatment [20]. While interesting, studies like these were excluded from this review as they draw from polygenic disease risk to predict drug outcomes, rather than from pharmacogenetic associations with the drug outcome. Articles repurposing phenotype-derived polygenic risk models for drug outcome prediction were similarly excluded. For example, Helmstaedter et al. sought to predict levetiracetam-induced behavioural side-effects using SNPs that predisposed individuals to impulsive, reactive, or aggressive behaviours [21]. As this polygenic model does not incorporate pharmacogenetic variants involved in levetiracetam-induced outcomes, it was not considered a pharmacogenetic model. This is not to say these types of polygenic models are not useful in the prediction of drug outcomes, but simply that they fall outside the scope of the current review. Additionally, these studies were recently reviewed by Johnson et al., so this work sought instead to take a more focused approach in evaluating polygenic models encompassing pharmacogenetic variants only [19].
2.2. Search Details
Liberal search criteria were applied in order to capture all relevant articles. For the purposes of this review, a polygenic model was broadly defined as any model or score encompassing pharmacogenetic variants at more than one genetic locus used to stratify patients by genetic risk. Both weighted and unweighted models derived from candidate gene or genome-wide associations were included. Any specific drug-related outcomes were included, such as drug-dosing, therapeutic drug response, or drug-induced adverse effects. Studies that did not investigate pharmacological treatments (e.g., surgical procedures, supplementation, radiation therapy) were excluded. Additionally, studies that did not examine a specific drug (e.g., investigating a chemotherapeutic regimen rather than a specific chemotherapeutic agent) were excluded as it is not possible to associate pharmacogenetic findings to a specific drug-related phenotype. Studies repurposing disease-derived or phenotype-derived polygenic scores to predict drug-related outcomes (e.g., schizophrenia-derived polygenic risk score used to predict lurasidone response) were also excluded, as this was not considered a polygenic model developed using pharmacogenetic variants.
A librarian specializing in medical genetics research was consulted to help construct the search strategy. This was done beginning with MeSH terms, followed by key-words and variations. This strategy was further refined upon review of initial search results in order to ensure all relevant papers were being captured by the search. For example, search terms pertaining to ‘personalized medicine’ were included in the search strategy as some pharmacogenetic studies were filed under this concept within the databases and not necessarily within the ‘pharmacogenetics’ search term. The final search strategy consisted of terms pertaining to “pharmacogenomics”, “pharmacogenetics”, “personalized medicine”, and “polygenic model”. The full search strategy is shown in Figure S1. The search was conducted in MEDLINE and EMBASE using the OVID interface from 1946 to 27 July 2021 for articles that described the development or validation of a polygenic model in human subjects to predict any drug outcomes.
2.3. Study Selection
Study screening was performed by two independent reviewers (A.S. and S.A.) in order to minimize bias and retrieve all relevant records pertaining to the research question. Articles were screened for relevancy by their title and abstract, followed by full-text review. To begin the title and abstract screening process, reviewers screened 20 articles for inclusion in full-text review. Conflicts were resolved through discussion between reviewers until a minimum inter-rater reliability of α = 0.8 was reached. Articles selected for full review included English language original studies containing a polygenic model used to predict drug outcomes. Conference abstracts, case reports, editorials, notes, meta-analyses, and review articles were not included for full-text review.
2.4. Data Extraction
Data extraction was performed by a single reviewer (AS). A standard data extraction sheet was created and piloted on 10 articles, and necessary changes were made to the form before applying it to the full list of included papers. Additional articles were excluded during this process as they were not found to meet all inclusion criteria upon detailed review. Names of the lead authors were extracted, as well as the year of publication. Drugs were classified using the LexiComp database according to their pharmacological category [22]. Details of the drug outcome under investigation were extracted and categorized according to safety, efficacy, or dosing predictions. The method of selecting pharmacogenetic variants for consideration into model development was collected and categorized according to candidate-gene or genome-wide association methods. Details of the model training and validation cohorts were extracted, including population details, as well as number of patients in all study cohorts. Development cohorts were defined as populations used to develop the original score or model, and validation cohorts were defined as any independent population used to test the model’s predictive capabilities. Details of model performance measures were also extracted where available. In cases where the pharmacogenetic prediction model was compared to clinical prediction models, performance measures of the comparison were extracted. If the model was independently validated in an external cohort, predictive performance of the model in this independent population was also extracted. There is currently no risk of bias assessment tool for polygenic model reviews, thus this could not be formally assessed.
2.5. Synthesis of Results
Figures, plots, and measures of central tendency used to summarize the included articles were conducted in RStudio Version 1.3.959 for MacOS.
3. Results
3.1. Overview of Included Articles
The initial literature search conducted in MEDLINE and EMBASE identified 5132 articles. After removal of 514 duplicates, 4618 articles remained for title and abstract screening. From these, 4259 irrelevant studies were excluded by two independent investigators, leaving 359 reports to be extracted for full-text review. Following full-text screening, 100 articles were initially included. During detailed data extraction, an additional 11 reports were excluded as they were found not to include a pharmacogenetic model with multiple genes, leaving 89 papers for inclusion in the systematic review (Figure 1). Full details on included studies can be found in Table S1.
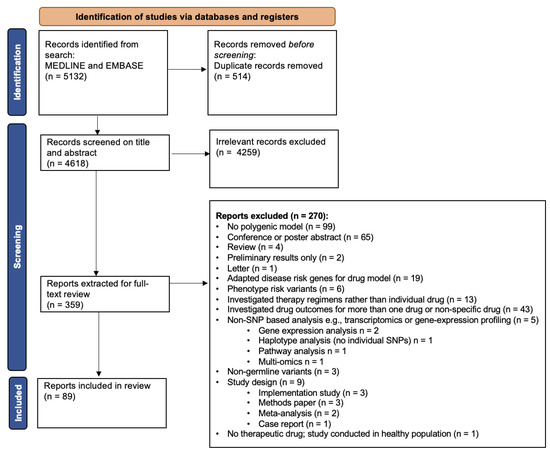
Figure 1.
CONSORT flow diagram of articles screened and included in the final review. Flow chart adapted from an example in Page et al. (2021) [23].
Included papers were published on or before the search date (27 July 2021). The vast majority of drugs for which polygenic predictive models are developed fall under anticoagulants (n = 32, 36.0%) or antineoplastic agents (n = 22, 24.7%). Of the anticoagulants studied, all were vitamin K antagonists. Drug outcomes under investigation were categorized under three main categories: drug safety (i.e., adverse drug reactions), drug dosing requirements (including drug exposure prediction), and drug effectiveness. Of these, drug dosing requirements was the most common outcome under investigation when developing polygenic prediction models (n = 33, 37.1%) [18,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55], followed by drug safety (n = 32, 36.0%) [56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87] and drug effectiveness (n = 24, 27.0%) [88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111]. The vast majority of studies investigating dosing requirements were conducted in regard to anticoagulant therapy, whereas the majority of drug safety studies were conducted in antineoplastics. A summary of investigated drug outcomes stratified by drug class can be seen in Figure 2.
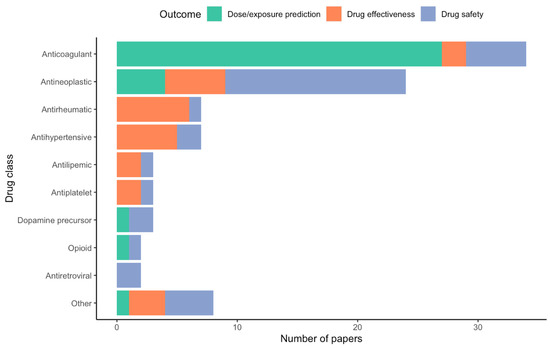
Figure 2.
Number of articles included in the review grouped by drug class and investigated drug outcome. The “Other” category is comprised of Sufonylurea antidiabetic (n = 1), Angiogenesis inhibitor (n = 1), 5-Aminosalicylic acid derivative (n = 1), Beta2 agonist (n = 1), Antifungal agent (n = 1), Antitubercular agent (n = 1), Immunosuppressant agent (n = 1), and Immune globulin (n = 1).
3.2. Method of Gene-Selection for Developing a Polygenic Model Predicting Drug Outcomes
74 of the 89 included studies (83.1%) used a candidate-gene approach when choosing SNPs for inclusion in a multi-pharmacogenetic prediction model. Only 11 studies (12.4%) performed a genome-wide or exome-wide association study to identify pharmacogenetic variants for model development [49,65,69,70,76,86,95,100,102,107,108]. An additional 4 studies (4.5%) aimed to validate a previously published polygenic model [87,109,110,111]. The preference for the candidate-gene approach in these articles may be explained by several factors. Candidate gene analyses are simpler to run and more cost-effective to perform [112,113]. Additionally, sample size constraints remain a challenge in pharmacogenomics research [113]. The median sample size for model development cohorts was 269 patients which would generally be underpowered to accurately estimate allele effect-sizes in a genome-wide study design [7].
Among the candidate-gene approaches, a variety of rationales were used for selection of candidate genes for model development. The most popular method was through literature search to identify variants previously associated with the drug outcome of interest or variants with functional relevance to the drug’s pharmacokinetic or pharmacodynamic pathways. Only two studies explicitly incorporated evidence-threshold criteria in the selection of candidate SNPs. The study by Palles et al. developing a prediction model for capecitabine-induced toxicities used a statistical evidence threshold to select variants associated with the drug outcome in studies of ≥500 patients with an OR/HR of ≥1.5 [75]. Another study by Leusink et al. examining statin-induced cholesterol lowering chose candidate SNPs for model development based on SNPs previously reaching genome-wide significance and replicated in at least one other study for the same drug outcome [104]. For pharmacogenetic models developed using candidate-SNPs, a range of 2 to 60 SNPs were incorporated into the predictive model.
Among the GWA studies, all studies set a p-value threshold for choosing SNPs to include in model development. Most studies set this threshold a priori, whereas two studies by Suzuki et al. examining mesalamine allergy and Lanfear et al. examining overall-survival in patients on β-blocker therapy used varying p-value thresholds to maximize model performance [70,114]. Some studies, like that by Sordillo et al. investigating albuterol response in children with asthma, set a modest p-value threshold (p < 0.001) but also incorporated functional criteria in SNP-selection by restricting SNPs to those whose predicted functional consequence exceeded 10 on the Combined Annotation Dependent Depletion (CADD) scale [102]. Pharmacogenetic models developed using GWAS included between 5 and 610 SNPs into the predictive pharmacogenetic model.
3.3. Overview of Methods Used to Develop Polygenic Predictions Models in Pharmacogenomics
Once pharmacogenetic SNPs were selected for inclusion into a polygenic prediction model, a variety of statistical methods were employed for the development of the models. These include regression-based methods, such as linear, logistic, or Cox proportional hazards regression analyses, and machine learning methods. Machine learning methods varied widely from more common techniques like random forest analyses to newly developed machine learning algorithms. The details of each of the different machine learning methods are beyond the scope of this review, and papers were broadly classified as using regression-based modelling (n = 68, 76.4%) or machine learning modelling (n = 11, 12.4%). A subset of papers used neither of these, relying instead on pharmacokinetic modelling techniques to create a polygenic prediction model (2 papers, 2.2%) [28,88], Baeysian probability modelling (1 paper, 1.1%) [50], or simply binned patients according to their genotype-category without applying any statistical modelling (7 papers, 7.9%) [42,45,52,59,75,93,105]. No difference was observed between the model development method and the model’s performance (p = 0.09). The methods for SNP-selection and modelling technique are summarized in Table 1.

Table 1.
Summary of sample and methods used for developing polygenic prediction models in pharmacogenomics research.
3.4. Performance of Polygenic Models in Pharmacogenomics Research
Given the variability in methodologies used to develop polygenic prediction models, it is unsurprising that the same heterogeneity exists for measuring model performance. Methods for assessing model performance included plotting receiver operating characteristic (ROC) curves and calculating area under the curve (AUC) as a measure of model discrimination, R2 measures of predictive accuracy, model calibration as measured by the Hosmer-Lemeshow goodness-of-fit test, sensitivity and specificity, positive- and negative-predictive values, mean absolute error, and Pearson correlations (for continuous outcomes only). Some studies did not formally evaluate model performance; rather, patients were binned into risk groups based on polygenic model score and association with the drug outcome was compared between groups. Given the variance in reporting of model performance, direct comparisons could not be drawn between models across different studies.
Instead, performance results were interpreted within the context of each individual study by examining (1) whether the polygenic model was successfully associated with the drug outcome of interest, and (2) whether it was able to improve predictions beyond clinical models. Nearly all included studies that developed a model (n = 83, 93.3%) identified a significant association between the drug outcome of interest and the pharmacogenetic variants incorporated into the model. However, less than half of these studies (n = 42, 47.2%) compared the polygenic model against clinical predictors. Comparisons against clinical predictors are used to demonstrate the added utility of pharmacogenetics beyond clinical factors alone in predicting drug outcomes [115]. Of the studies that did make this comparison, 73.8% showed a significant improvement of the polygenic model over a clinical model. A summary of models reporting significant polygenic associations and improvement over clinical models is shown in Figure 3.
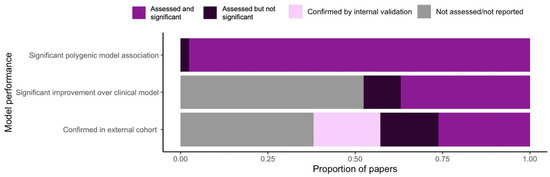
Figure 3.
Summary of performance and validation of polygenic models for predicting drug outcomes.
3.5. Validating the Performance of Polygenic Models
Over half the included papers (n = 56, 63.0%) included some form of model validation in their analysis or were validated in a future study. However, only 36 (40.4%) models were tested in an independent cohort for external validity. As mentioned previously, secondary cohorts of patients treated with the same drug may not be readily available due to sample size constraints. In these cases, some studies (n = 16, 18.0%) performed internal validation using cross-validation or internal bootstrap samples to validate their model. Expectedly, internal validation of polygenic prediction models was far more successful than external validation. Where all internally validated models reported successful validation with only a slight reduction in performance, over one third (n = 14, 38.9%) of externally validated models did not validate successfully in an independent patient population. Model validation is also summarized in Figure 3.
A very small subset of papers (n = 4, 4.5%) was dedicated solely to the independent validation of a previously developed polygenic model. This is in line with the trend in scientific research which has historically favored discovery over replication for publication and explains why most studies aimed to created their own polygenic model rather than validate an existing one [116].
4. Discussion
4.1. Drug Outcomes Investigated
A wide range of therapeutic classes have been investigated among the included studies in the development of polygenic prediction models (Figure 2). However, the studies reviewed were heavily dominated by anticoagulant (n = 32, 36.0%) and antineoplastic (n = 22, 24.7%) outcome prediction. Historically, coumarin anticoagulants were extensively studied in the context of pharmacogenomics research due to the widespread prescription of warfarin for the prophylaxis and treatment of venous thromboembolism and other cardiac conditions [117,118,119]. Due to the narrow therapeutic index and high interindividual variability in dosing requirements, ability to predict a patient’s optimal warfarin dose is crucial for avoiding serious adverse drug reactions [120,121]. Prior to any genome-wide studies, researchers and clinicians already suspected that up to 50% of this variability could be explained by patient-specific factors such as age, body mass index, and genetics [14]. Up to one-third of this variability has been associated with variations in the main metabolizer enzyme for coumarin anticoagulants, CYP2C9, and the primary drug target, vitamin K epoxide reductase complex I (VKORC1) [122]. This prompted the FDA to include pharmacogenetic information on the warfarin drug label, and the International Warfarin Pharmacogenetics Consortium to produce a standard drug-dosing algorithm for warfarin prescription based on genetic information [18,123]. The extensive research on warfarin pharmacogenetics makes it a compelling case study for the polygenic nature of individual drug response, as well as how the use of pharmacogenetic testing can optimize drug outcome predictions.
The pharmacogenetics of anti-cancer therapies have also been extensively investigated. The potent pharmacological agents used to prolong life in cancer can result in severe adverse drug reactions which disproportionately affect cancer patients, with up to 74.3% of hospitalized oncology patients experiencing one or more adverse drug reaction during their stay [124]. As advancements in cancer therapy have improved patient survival, increasing attention has been given to the life-altering and life-threatening adverse effects of chemotherapy [125,126,127]. It is, therefore, unsurprising that the majority of polygenic risk models in cancer therapeutics were developed to predict individual susceptibility to chemotherapy-related adverse drug reactions (Figure 2).
4.2. Methods for Polygenic Model Development
In the context of this review, the term ‘polygenic’ was not restricted to the classical definition of “a sum of genome-wide genotypes” [8]. Instead, the term ‘polygenic’ was broadly defined as any pharmacogenetic prediction score or model that encompassed more than one genetic locus in order to also capture pharmacogenetic models not developed from genome-wide studies. Nearly all studies included in this review took a candidate-gene approach when choosing pharmacogenetic SNPs to incorporate into a polygenic prediction model which are widely regarded as inferior to GWA studies due to their hypothesis-driven nature. Linskey and colleagues identified that 94% of genes discovered in pharmacogenomic GWA studies are novel and not previously included in candidate gene studies [128]. This demonstrates the gap in our current understanding of drug pathways and emphasizes the need to shift pharmacogenomic research towards agnostic genome-wide study designs.
The current preference for candidate-gene studies may be explained by the small average sample sizes available in the included articles (median n = 269) which would generally be considered underpowered for genome-wide analyses [7]. However, pharmacogenetic variants tend to have larger effects sizes compared to variants associated with other complex traits [129]. While smaller samples may suffice for detection of these larger effect sizes, pharmacogenetic associations of modest effect involved in complex drug pathways may still be missed [113]. This highlights a common challenge within pharmacogenomics research of recruiting sufficiently large samples of uniformly treated patients to perform GWA studies [7,19,113]. This has led many researchers to leverage GWASs derived from large cohorts of related disease phenotypes in the development of polygenic models to predict drug outcome [19]. As mentioned, these studies fell outside the scope of this review as they failed to include pharmacogenetic variants. GWA studies also present additional challenges that may have contributed to the preference for candidate-gene approaches among the included articles. Due to their large scale, GWA studies are often more complex, more time-consuming, and more expensive to run as they require statistical experts familiar with genomic analyses, higher computing power, and specialized genetic analysis software [112].
Nearly all included studies employed regression-based statistical modelling techniques to develop the polygenic prediction models, with only 11 (12.4%) papers using machine learning techniques. Currently, there is not one methodology that produces the best model across all contexts or drug outcomes; rather, it appears that each drug outcome is assessed independently based on the phenotype and study population to determine the most suitable modelling method [115,130]. This is in line with findings from the current review, where no difference was observed between the method used to create the model and the model’s performance (p = 0.09).
This suggests that it is perhaps the data used to create the model which has more impact on model performance than the method of model creation [131,132]. For instance, Perini et al., found that a warfarin dosing algorithm developed in a Brazilian population outperformed previous models developed in European populations when applied to Brazilian patients [46]. This is unsurprising given the genetic differences between ancestries. Variant frequency and linkage disequilibrium patterns can vary widely between populations, which often translates to poor performance of polygenic models applied to patients who are different from the input data [133,134,135]. Another study in warfarin dose prediction compared the performance of various models for predicting dosing requirements in children [47]. This study found that the model generated in a pediatric population outperformed those that adapted warfarin dosing models constructed in adults for use in children [47]. This demonstrates that a model performs best within the population for which it was developed, particularly when populations have differing pharmacokinetic profiles [136].
Phenotypic characterization also presents a unique challenge within pharmacogenomic research as many drug outcomes are difficult to measure quantitatively [14]. For example, cisplatin-induced hearing loss is a common adverse drug reaction resulting from cisplatin chemotherapy [137,138,139]. Many pharmacogenomic studies have been conducted to explain the interindividual variability of this adverse outcome, but results are inconsistently replicated [140]. This may be partially explained by the different scales used to grade hearing loss which results in the same patient being assigned into different phenotypic categories depending on the grading criteria used [137,141,142,143,144,145]. Such discrepant phenotyping may result in different polygenic models being constructed depending on the definition of the drug outcome.
There is a wide variety of methods for generating polygenic models in pharmacogenomics research and this diversity continues to increase as different mechanisms arise to overcome challenges in modelling complex drug-related data [115]. This presents a challenge as each drug outcome may have multiple polygenic models with little guidance in choosing the ‘correct’ model to implement clinically. Additionally, polygenic models constructed using more complicated or abstract techniques may face additional barriers toward clinical implementation [131,132,146]. For example, due to the data-driven nature of machine learning methods, learning algorithms are often perceived as a “black box”, manipulating data in unknown ways to generate predictions. Due to the limited interpretability of algorithm results, clinicians and practitioners may have difficulty trusting a model that is not easily explained by current medical evidence [146]. This illustrates a need for data scientists and clinicians to work together in early stages of model development in order to create polygenic prediction models that are clinically useful and interpretable by its intended end-users.
4.3. Model Performance
All except for two studies in the current review found significant polygenic associations between the studied polygenic model with the drug outcome of interest. This is in contrast to findings from a review published by Johnson et al. in 2021 where more than half the included studies did not find a significant association between the polygenic risk score and the drug outcome of interest [19]. This difference may be attributed to the fact that variants incorporated into many of the prediction models reviewed by Johnson et al. were disease-related rather than drug-related, and hence did not capture the true pharmacogenetic landscape of the drug outcome under investigation [19]. Articles included in the present review comprised of pharmacogenetic variants previously found to be in direct association with drug outcomes, or with established functional relevance in the drug’s biotransformation pathway. This may explain why the overwhelming majority of studies in this review found a statistically significant association between polygenic models and the drug outcomes. These findings suggest that disease-associated variants cannot always substitute for true pharmacogenetic associations. Pharmacological agents form complex interactions with biological systems through various pharmacokinetic and pharmacodynamic pathways that extend beyond disease mechanisms [147]. Thus, the most robust polygenic models for predicting drug response are those constructed using pharmacogenetic variants. However, these results should be interpreted with caution as studies failing to show a statistically significant association between pharmacogenetic models and investigated drug outcomes may be more likely to remain unpublished [148].
While nearly all studies were able to show a significant genetic association between their polygenic models and drug outcomes, far fewer demonstrated that the inclusion of pharmacogenetic information significantly improved predictions beyond clinical factors alone. Less than half (n = 42, 47.2%) the included studies formally compared polygenic versus clinical models for predicting drug outcomes. Of the models that did draw this comparison 73.8% (n = 31 out of 42) showed significant improvement over clinical models with the addition of pharmacogenetic factors, suggesting that pharmacogenetics have the potential to improve prediction of drug outcomes over clinical models alone. However, this should also be interpreted with caution due to the low proportion of studies that reported the predictive performance of clinical versus pharmacogenetic models. It is possible that negative results failing to demonstrate improvement of polygenic models over clinical models are less likely to be reported [148]. This may also be due to the lack of established clinical prediction tools against which to compare pharmacogenetic models. Unlike for predicting disease risk, validated clinical prediction tools often do not exist for predicting drug outcomes. Nevertheless, clinical factors have been associated with many drug outcomes of interest. For example, younger-aged children tend to be more at-risk for chemotherapy-induced adverse reactions and body mass index is a well-established predictor for warfarin dosing requirements [149,150,151,152]. Comparisons between these clinical factors and polygenic models are crucial to show clinicians and stakeholders how pharmacogenetics can be used in conjunction with clinical information to result in more effective, individualized therapy [153,154,155,156]. Reporting the extent to which a polygenic model is able to improve (or not) upon clinical predictions where available is likely to play an important role in the implementation of pharmacogenetic testing.
The diversity that exists within model development methods also exists within the reporting of predictive performance. This variability makes comparison and evaluation of polygenic models challenging when trying to decide on the ‘best’ model for use in patients [11]. For instance, the area under the curve (AUC) is the most frequently used metric of a model’s discriminative ability, but it has also been criticized as lacking in other predictive aspects [157]. This has led some authors to instead report metrics of calibration, mean-average-error, or percent variability explained (among many others). Recommendations for reporting practices and guidelines for polygenic model development have been published in the context of disease prediction but are not routinely followed [8,158,159,160]. This inconsistency is apparent in the vast array of performance measures reported among the included studies in this article, and the same trend was observed in a recent review of polygenic risk scores [19]. Improving adherence to standardized reporting guidelines would facilitate comparisons between polygenic prediction models and allow more straightforward evaluation of model performance. Additionally, there are currently no reporting guidelines that are specific to pharmacogenetic polygenic models. Thus, it remains to be seen whether guidelines for disease polygenic models are applicable to pharmacogenetic models, and if so, consensus must be reached on the one(s) to follow in order to facilitate cross-study comparisons.
4.4. Model Validation
In order for any prediction model to be implemented, validation of the model must occur in order to demonstrate its predictive performance. In the current review, over one-third of studies (n = 33, 37.1%) did not include any obvious form of validation. That is, a polygenic model was fit to the data without testing the validity of genotype-based groupings or predictions. n = 16, (18.0%) performed internal validation only using bootstrap or other re-sampling techniques. However, it is widely accepted that it is not sufficient to demonstrate good model performance in the development sample only [161]. In order to demonstrate generalizability, it is essential to confirm that a model maintains good prediction in a different set of individuals than were used for model creation [162]. In this review, a low proportion of articles (n = 36, 40.4%) validated the polygenic model in an independent test sample. Only n = 4 (4.5%) of the articles were focused solely on conducting an external validation of an existing polygenic prediction model. This low number may be explained by the tendency to preferentially produce novel research rather than attempt to replicate previously published results [116,163,164,165,166,167]. Often, especially until more recent years, publication preference has been given to novel findings [116,163,164,165,166,167]. However, replication of polygenic models for predicting drug outcomes is key to demonstrating their generalizability across patient populations. Generalizability of model predictions has been particularly challenging in the development of polygenic prediction models, with a drop in model performance often observed when applied to a new patient population [135,168,169,170]. This trend is observed in the present study where over one-third of externally validated models failed to predict the drug outcome in an independent cohort (Figure 3.). Part of this challenge reflects a larger bias in genetic research which has primarily been conducted in European populations (Table S1) [113]. As a result, many of these genetic findings are not applicable to populations of different ancestries. Recently, suggestions have been made for reweighting or adjusting models when applied to different populations, but ultimately, there is a need to increase patient-diversity in genetic studies [171]. It has been previously demonstrated that polygenic models developed in more diverse samples have improved generalizability and improved performance when applied to external cohorts of different populations [134]. Thus, improving diversity in pharmacogenetic research is an essential step in creating polygenic models that are widely applicable. Fostering international research collaborations and the formation of large consortia comprised of genetically diverse patients would allow for improved generalizability of pharmacogenetic predictions and more widespread applications of polygenic models.
4.5. Study Limitations and Future Directions
This work has several limitations. As discussed previously, the scope of the current study was limited to polygenic models constructed from pharmacogenetic variants only and excluded those derived from disease or phenotype GWAS data. As such, direct comparisons could not be drawn between these different models. Future studies may consider performing a larger-scale review which directly compares these different models, particularly where both are available for the same drug outcomes. This study also excluded polygenic models constructed for multi-drug regimens and thus the results cannot be generalized to drug outcomes resulting from the combined effect of multiple pharmacotherapies. An additional limitation is the exclusion of any non-English language articles as this may have introduced bias into the current study and caused some evidence to be missed. Finally, due to the heterogeneity in reporting of model results, no assessment of publication bias was conducted. As mentioned, negative results are less likely to be reported and thus, the effects of publication bias on the results of the current review cannot be ruled out [148]. This highlights the need to establish clear reporting guidelines for polygenic models predicting drug outcomes, as well as the need to report negative findings to reduce publication bias. Another important consideration in future work is the integration of multiple gene effects (polygenic models) into clinical practice guidelines for pharmacogenetic testing. Currently, clinical practice recommendations for pharmacogenetic testing are predominantly made on a per-gene basis [172,173]. Clear guidelines on clinical interpretation of pharmacogenetic results that combine multiple variants are needed.
In conclusion, the development of polygenic models for predicting drug outcomes is an emerging field with the potential to improve predictions for individual patient response to pharmacological therapy. However, to facilitate advancements in this area of research, consensus is needed surrounding the reporting of model development methods and model performance measures. Additionally, increasing diversity in study populations for polygenic model development can lead to improved generalizability of model predictions and demonstrate clinical utility in a broader group of patients.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jpm12091394/s1, Figure S1: Full search strategy; Table S1: Summary of included articles.
Author Contributions
Conceptualization, A.S. and B.C.C.; methodology, A.S.; data curation, A.S. and S.J.A.; investigation, A.S.; visualization, A.S.; writing—original draft preparation, A.S.; writing—review and editing, S.J.A., S.R.R., C.J.D.R. and B.C.C.; supervision, B.C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was supported in part by a grant from Genome Canada, Genome British Columbia (funding #272PGX) and the Canadian Institutes of Health Research (CIHR) (funding #GP1-155872) through the Large Scale Applied Research Project Competition. Additional support has been provided by British Columbia’s Provincial Health Services Authority (PHSA); BC Children’s Hospital Foundation; Health Canada; Illumina, Thermo Fisher. Angela Siemens was also supported by CIHR’s MSc Studentship and Drug Safety and Effectiveness Cross-Disciplinary Training Programs.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Template data collection forms and analytic code used in this study is available from the authors upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fisher, R.A. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 1918, 52, 399–433. [Google Scholar] [CrossRef]
- Zeggini, E.; Gloyn, A.L.; Barton, A.C.; Wain, L.V. Translational genomics and precision medicine: Moving from the lab to the clinic. Science 2019, 365, 1409–1413. [Google Scholar] [CrossRef]
- Wray, N.R.; Goddard, M.E.; Visscher, P.M. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007, 17, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Park, B.K. Genetic susceptibility to adverse drug reactions. Trends Pharmacol. Sci. 2001, 22, 298–305. [Google Scholar] [CrossRef]
- Alfirevic, A.; Pirmohamed, M. Genomics of Adverse Drug Reactions. Trends Pharmacol. Sci. 2017, 38, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; Macarthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.H.; O’Reilly, P.F. A guide to performing Polygenic Risk Score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Guinan, K.; Beauchemin, C.; Tremblay, J.; Chalmers, J.; Woodward, M.; Tahir, M.R.; Hamet, P.; Lachaine, J. Economic Evaluation of a New Polygenic Risk Score to Predict Nephropathy in Adult Patients with Type 2 Diabetes. Can. J. Diabetes 2021, 45, 129–136. [Google Scholar] [CrossRef]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Forgetta, V.; Keller-Baruch, J.; Forest, M.; Durand, A.; Bhatnagar, S.; Kemp, J.P.; Nethander, M.; Evans, D.; Morris, J.A.; Kiel, D.P.; et al. Development of a polygenic risk score to improve screening for fracture risk: A genetic risk prediction study. PLoS Med. 2020, 17, e1003152. [Google Scholar] [CrossRef]
- Auwerx, C.; Sadler, M.C.; Reymond, A.; Kutalik, Z. From Pharmacogenetics to Pharmaco-Omics:Milestones and Future Directions. Hum. Genet. Genom. Adv. 2022, 3, 100100. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K. Genome-wide association studies in pharmacogenomics. Nat. Rev. Genet. 2010, 11, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.M.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Johnson, J.A.; Langaee, T.Y.; Feng, H.; Stanaway, I.B.; Schwarz, U.I.; Ritchie, M.D.; Stein, C.M.; Roden, D.M.; Smith, J.D.; et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008, 112, 1022–1027. [Google Scholar] [CrossRef]
- Takeuchi, F.; McGinnis, R.; Bourgeois, S.; Barnes, C.; Eriksson, N.; Soranzo, N.; Whittaker, P.; Ranganath, V.; Kumanduri, V.; McLaren, W.; et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009, 5, e1000433. [Google Scholar] [CrossRef]
- The International Warfarin Pharmacogenetics Consortium. Estimation of the Warfarin Dose with Clinical and Pharmacogenetic Data. N. Engl. J. Med. 2009, 360, 753–764. [Google Scholar] [CrossRef]
- Johnson, D.; Wilke, M.A.P.; Lyle, S.M.; Kowalec, K.; Jorgensen, A.; Wright, G.E.B.; Drögemöller, B.I. A Systematic Review and Analysis of the Use of Polygenic Scores in Pharmacogenomics. Clin. Pharmacol. Ther. 2021, 111, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yoshikawa, A.; Brennan, M.D.; Ramsey, T.L.; Meltzer, H.Y. Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr. Res. 2018, 192, 194–204. [Google Scholar] [CrossRef]
- Helmstaedter, C.; Mihov, Y.; Toliat, M.R.; Thiele, H.; Nuernberg, P.; Schoch, S.; Surges, R.; Elger, C.E.; Kunz, W.S.; Hurlemann, R. Genetic variation in dopaminergic activity is associated with the risk for psychiatric side effects of levetiracetam. Epilepsia 2013, 54, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lexicomp. Available online: https://online-lexi-com.eu1.proxy.openathens.net/lco/action/home?siteid=2 (accessed on 17 May 2022).
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shennan, M.; Reynolds, K.K.; Johnson, N.A.; Herrnberger, M.R.; Valdes, R. Estimation of warfarin maintenance dose based on VKORC1 (−1639 G > A) and CYP2C9 genotypes. Clin. Chem. 2007, 53, 1199–1205. [Google Scholar] [CrossRef]
- Sconce, E.A.; Khan, T.I.; Wynne, H.A.; Avery, P.; Monkhouse, L.; King, B.P.; Wood, P.; Kesteven, P.; Daly, A.K.; Kamali, F. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: Proposal for a new dosing regimen. Blood 2005, 106, 2329–2333. [Google Scholar] [CrossRef]
- Gage, B.F.; Eby, C.; Milligan, P.E.; Banet, G.A.; Duncan, J.R. Use of pharmacogenetics and clinical factors to predict the maintenance dose of warfarin. Thromb. Haemost. 2004, 91, 87–94. [Google Scholar] [CrossRef]
- Nowak-Gottl, U.; Dietrich, K.; Schaffranek, D.; Eldin, N.S.; Yasui, Y.; Geisen, C.; Mitchell, L.G. In pediatric patients, age has more impact on dosing of vitamin K antagonists than VKORC1 or CYP2C9 genotypes. Blood 2010, 116, 6101–6105. [Google Scholar] [CrossRef]
- Lala, M.; Burckart, G.J.; Takao, C.M.; Pravica, V.; Momper, J.D.; Gobburu, J.V.S. Genetics-based pediatric warfarin dosage regimen derived using pharmacometric bridging. J. Pediatr. Pharmacol. Ther. 2013, 18, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.; Amstutz, U.; Hildebrand, C.; Rassekh, S.R.; Hosking, M.; Neville, K.; Leeder, J.S.; Hayden, M.R.; Ross, C.J.; Carleton, B.C. VKORC1 and CYP2C9 genotypes are predictors of warfarin-related outcomes in children. Pediatr. Blood Cancer 2014, 61, 1055–1062. [Google Scholar] [CrossRef]
- Vear, S.I.; Ayers, G.D.; van Driest, S.L.; Sidonio, R.F.; Stein, C.M.; Ho, R.H. The impact of age and CYP2C9 and VKORC1 variants on stable warfarin dose in the paediatric population. Br. J. Haematol. 2014, 165, 832–835. [Google Scholar] [CrossRef]
- Nguyen, N.; Anley, P.; Yu, M.Y.; Zhang, G.; Thompson, A.A.; Jennings, L.J. Genetic and clinical determinants influencing warfarin dosing in children with heart disease. Pediatr. Cardiol. 2013, 34, 984–990. [Google Scholar] [CrossRef]
- Moreau, C.; Bajolle, F.; Siguret, V.; Lasne, D.; Golmard, J.L.; Elie, C.; Beaune, P.; Cheurfi, R.; Bonnet, D.; Loriot, M.A. Vitamin K antagonists in children with heart disease: Height and VKORC1 genotype are the main determinants of the warfarin dose requirement. Blood 2012, 119, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Biss, T.T.; Avery, P.J.; Brandao, L.R.; Chalmers, E.A.; Williams, M.D.; Grainger, J.D.; Leathart, J.B.S.; Hanley, J.P.; Daly, A.K.; Kamali, F. VKORC1 and CYP2C9 genotype and patient characteristics explain a large proportion of the variability in warfarin dose requirement among children. Blood 2012, 119, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Wadelius, M.; Chen, L.Y.; Bumpstead, S.; Ghori, J.; Bentley, D.; McGinnis, R.; Deloukas, P.; Eriksson, N.; Wadelius, C. Association of warfarin dose with genes involved in its action and metabolism. Hum. Genet. 2007, 121, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Limdi, N.A.; Arnett, D.K.; McGwin, G.; Goldstein, J.A.; Beasley, T.M.; Adler, B.K.; Acton, R.T. Influence of CYP2C9 and VKORC1 on warfarin dose, anticoagulation attainment and maintenance among European-Americans and African-Americans. Pharmacogenomics 2008, 9, 511–526. [Google Scholar] [CrossRef]
- Spreafico, M.; Mannucci, P.M.; Peyvandi, F.; Lodigiani, C.; Rota, L.L.; van Leeuwen, Y.; Rosendaal, F.R.; Pizzotti, D. Effects of CYP2C9 and VKORC1 on INR variations and dose requirements during initial phase of anticoagulant therapy. Pharmacogenomics 2008, 9, 1237–1250. [Google Scholar] [CrossRef]
- Kurnik, D.; Qasem, H.; Sominski, S.; Halkin, H.; Gak, E.; Loebstein, R.; Li, C.; Stein, C. Effect of the VKORC1 ASP36TYR variant on warfarin dose requirement and pharmacogenetic dose prediction models. Clin. Pharmacol. Ther. 2012, 91 (Suppl. S1), S96–S135. [Google Scholar] [CrossRef]
- Teh, L.K.; Langmia, I.M.; Fazleen Haslinda, M.H.; Salleh, M.Z.; Ngow, H.A.; Roziah, M.J.; Harun, R.; Zakaria, Z.A. Clinical relevance of VKORC1 (G-1639A and C1173T) and CYP2C9*3 among patients on warfarin. J. Clin. Pharm. Ther. 2012, 37, 232–236. [Google Scholar] [CrossRef]
- Wang, D.; Wu, H.; Chong, J.; Lu, Y.; Yin, R.; Zhao, X.; Zhao, A.; Yang, J.; Chen, H.; Dai, D.P. Effects of rare CYP2C9 alleles on stable warfarin doses in Chinese Han patients with atrial fibrillation. Pharmacogenomics 2020, 21, 1021–1031. [Google Scholar] [CrossRef]
- Yoshida, K.; Nishizawa, D.; Ichinomiya, T.; Ichinohe, T.; Hayashida, M.; Fukuda K ichi Ikeda, K. Prediction formulas for individual opioid analgesic requirements based on genetic polymorphism analyses. PLoS ONE 2015, 10, e0116885. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tsunedomi, R.; Fujita, Y.; Otori, T.; Ohba, M.; Kawai, Y.; Hirata, H.; Matsumoto, H.; Haginaka, J.; Suzuki, S.; et al. Pharmacogenetics-based area-under-curve model can predict efficacy and adverse events from axitinib in individual patients with advanced renal cell carcinoma. Oncotarget 2018, 9, 17160–17170. [Google Scholar] [CrossRef]
- Verhoef, T.I.; Redekop, W.K.; Buikema, M.M.; Schalekamp, T.; Van Der Meer, F.J.M.; LE Cessie, S.; Wessels, J.A.M.; Van Schie, R.M.F.; DE Boer, A.; Teichert, M.; et al. Long-term anticoagulant effects of the CYP2C9 and VKORC1 genotypes in acenocoumarol users. J. Thromb. Haemost. 2012, 10, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Chen, Y.J.; Fu, X.; Jiang, B.; Zhang, Y. Evolutionary Ensemble Learning Algorithm to Modeling of Warfarin Dose Prediction for Chinese. IEEE J. Biomed. Health Inform. 2019, 23, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Wenying, S.; Lingyan, C.; Xiaoye, H.; Meimei, Z.; Wensheng, C.; Lei, M.; Xiaoyan, L.; Jianing, H.; Tingyuan, P.; Jia, L.; et al. Cytochrome P450 Genetic Variations Can Predict mRNA Expression, Cyclophosphamide 4-Hydroxylation, and Treatment Outcomes in Chinese Patients with Non-Hodgkin’s Lymphoma. J. Clin. Pharmacol. 2017, 57, 886–898. [Google Scholar] [CrossRef]
- Shahabi, P.; Scheinfeldt, L.B.; Lynch, D.E.; Schmidlen, T.J.; Perreault, S.; Keller, M.A.; Kasper, R.; Wawak, L.; Jarvis, J.P.; Gerry, N.P.; et al. An expanded pharmacogenomics warfarin dosing table with utility in generalised dosing guidance. Thromb. Haemost. 2016, 116, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Perini, J.A.; Struchiner, C.J.; Silva-Assuncao, E.; Santana, I.S.C.; Rangel, F.; Ojopi, E.B.; Dias-Neto, E.; Suarez-Kurtz, G. Pharmacogenetics of warfarin: Development of a dosing algorithm for brazilian patients. Clin. Pharmacol. Ther. 2008, 84, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Marek, E.; Momper, J.D.; Hines, R.N.; Takao, C.M.; Gill, J.C.; Pravica, V.; Gaedigk, A.; Burckart, G.J.; Neville, K.A. Prediction of Warfarin Dose in Pediatric Patients: An Evaluation of the Predictive Performance of Several Models. J. Pediatr. Pharmacol. Ther. 2016, 21, 224–232. [Google Scholar] [CrossRef]
- Grossi, E.; Podda, G.M.; Pugliano, M.; Gabba, S.; Verri, A.; Carpani, G.; Buscema, M.; Casazza, G.; Cattaneo, M. Prediction of optimal warfarin maintenance dose using advanced artificial neural networks. Pharmacogenomics 2014, 15, 29–37. [Google Scholar] [CrossRef]
- Eriksson, N.; Wallentin, L.; Berglund, L.; Axelsson, T.; Connolly, S.; Eikelboom, J.; Ezekowitz, M.; Oldgren, J.; Pare, G.; Reilly, P.; et al. Genetic determinants of warfarin maintenance dose and time in therapeutic treatment range: A RE-LY genomics substudy. Pharmacogenomics 2016, 17, 1425–1439. [Google Scholar] [CrossRef]
- De Graan, A.J.M.; Elens, L.; Smid, M.; Martens, J.W.; Sparreboom, A.; Nieuweboer, A.J.M.; Friberg, L.E.; Elbouazzaoui, S.; Wiemer, E.A.C.; van der Holt, B.; et al. A pharmacogenetic predictive model for paclitaxel clearance based on the DMET platform. Clin. Cancer Res. 2013, 19, 5210–5217. [Google Scholar] [CrossRef]
- Altmann, V.; Schumacher-Schuh, A.F.; Rieck, M.; Callegari-Jacques, S.M.; Rieder, C.R.M.; Hutz, M.H. Influence of genetic, biological and pharmacological factors on levodopa dose in Parkinson’s disease. Pharmacogenomics 2016, 17, 481–488. [Google Scholar] [CrossRef]
- Kurnik, D.; Qasim, H.; Sominsky, S.; Lubetsky, A.; Markovits, N.; Li, C.; Stein, C.M.; Halkin, H.; Gak, E.; Loebstein, R. Effect of the VKORC1 D36Y variant on warfarin dose requirement and pharmacogenetic dose prediction. Thromb. Haemost. 2012, 108, 781–788. [Google Scholar] [PubMed]
- Finkelman, B.S.; French, B.; Bershaw, L.; Brensinger, C.M.; Streiff, M.B.; Epstein, A.E.; Kimmel, S.E. Predicting prolonged dose titration in patients starting warfarin. Pharmacoepidemiol. Drug Saf. 2016, 25, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Anton, A.I.; Cerezo-Manchado, J.J.; Padilla, J.; Perez-Andreu, V.; Corral, J.; Vicente, V.; Roldan, V.; Gonzalez-Conejero, R. Novel associations of VKORC1 variants with higher acenocoumarol requirements. PLoS ONE 2013, 8, e64469. [Google Scholar] [CrossRef]
- Dapia, I.; Garcia, I.; Martinez, C.J.; Arias, P.; Guerra, P.; Diaz, L.; Garcia, A.; Ochoa, D.; Tenorio, J.; Ramirez, E.; et al. Prediction models for voriconazole pharmacokinetics based on pharmacogenetics: AN exploratory study in a Spanish population. Int. J. Antimicrob. Agents 2019, 54, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Schalekamp, T.; Brassé, B.P.; Roijers, J.F.; Chahid, Y.; van Geest-Daalderop, J.H.; de Vries-Goldschmeding, H.; van Wijk, E.M.; Egberts, A.C.; de Boer, A. VKORC1 and CYP2C9 genotypes and acenocoumarol anticoagulation status: Interaction between both genotypes affects overanticoagulation. Clin. Pharmacol. Ther. 2006, 80, 13–22. [Google Scholar] [CrossRef]
- Hoskins, J.M.; Rosner, G.L.; Ratain, M.J.; McLeod, H.L.; Innocenti, F. Pharmacodynamic genes do not influence risk of neutropenia in cancer patients treated with moderately high-dose irinotecan. Pharmacogenomics 2009, 10, 1139–1146. [Google Scholar] [CrossRef]
- Chang, T.J.; Liu, P.H.; Liang, Y.C.; Chang, Y.C.; Jiang Y der Li, H.Y.; Lo, M.T.; Chen, H.S.; Chuang, L.M. Genetic predisposition and nongenetic risk factors of thiazolidinedione- related edema in patients with type 2 diabetes. Pharm. Genom. 2011, 21, 829–836. [Google Scholar] [CrossRef]
- Vispo, E.; Cevik, M.; Rockstroh, J.K.; Barreiro, P.; Nelson, M.; Scourfield, A.; Boesecke, C.; Wasmuth, J.C.; Soriano, V. Genetic determinants of idiopathic noncirrhotic portal hypertension in HIV-infected patients. Clin. Infect. Dis. 2013, 56, 1117–1122. [Google Scholar] [CrossRef]
- Visscher, H.; Ross, C.J.D.; Rassekh, S.R.; Sandor, G.S.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatric Blood Cancer 2013, 60, 1375–1381. [Google Scholar] [CrossRef]
- Lima, A.; Bernardes, M.; Azevedo, R.; Monteiro, J.; Sousa, H.; Medeiros, R.; Seabra, V. SLC19A1, SLC46A1 and SLCO1B1 polymorphisms as predictors of methotrexate-related toxicity in Portuguese rheumatoid arthritis patients. Toxicol. Sci. 2014, 142, 196–209. [Google Scholar] [CrossRef]
- Custodio, A.; Moreno-Rubio, J.; Aparicio, J.; Gallego-Plazas, J.; Yaya, R.; Maurel, J.; Higuera, O.; Burgos, E.; Ramos, D.; Calatrava, A.; et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: A GEMCAD group study. Ann. Oncol. 2014, 25, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Ragia, G.; Tavridou, A.; Elens, L.; van Schaik, R.H.N.; Manolopoulos, V.G. CYP2C9*2 allele increases risk for hypoglycemia in POR*1/*1 type 2 diabetic patients treated with sulfonylureas. Exp. Clin. Endocrinol. Diabetes 2014, 122, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, Q.; Fu, S.; Xu, M.; Zhang, T.; Xie, C.; Feng, J.; Chen, J.; Zang, A.; Cai, Y.; et al. A novel genetic score model of UGT1A1 and TGFB pathway as predictor of severe irinotecan-related diarrhea in metastatic colorectal cancer patients. J. Cancer Res. Clin. Oncol. 2016, 142, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Chaix, M.A.; Parmar, N.; Kinnear, C.; Lafreniere-Roula, M.; Akinrinade, O.; Yao, R.; Miron, A.; Lam, E.; Meng, G.; Christie, A.; et al. Machine Learning Identifies Clinical and Genetic Factors Associated With Anthracycline Cardiotoxicity in Pediatric Cancer Survivors. JACC CardioOncology 2020, 2, 690–706. [Google Scholar] [CrossRef]
- Visscher, H.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; Carleton, B.C.; et al. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 2015, 16, 1065–1076. [Google Scholar] [CrossRef]
- Visscher, H.; Ross, C.J.D.; Rassekh, S.R.; Barhdadi, A.; Dubé, M.P.; Al-Saloos, H.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J. Clin. Oncol. 2012, 30, 1422–1428. [Google Scholar] [CrossRef]
- Vandell, A.G.; Walker, J.; Brown, K.S.; Zhang, G.; Lin, M.; Grosso, M.A.; Mercuri, M.F. Genetics and clinical response to warfarin and edoxaban in patients with venous thromboembolism. Heart 2017, 103, 1800–1805. [Google Scholar] [CrossRef]
- Vandell, A.G.; McDonough, C.W.; Gong, Y.; Langaee, T.Y.; Lucas, A.M.; Chapman, A.B.; Gums, J.G.; Beitelshees, A.L.; Bailey, K.R.; Johnson, R.J.; et al. Hydrochlorothiazide-induced hyperuricaemia in the pharmacogenomic evaluation of antihypertensive responses study. J. Intern. Med. 2014, 276, 486–497. [Google Scholar] [CrossRef]
- Suzuki, K.; Kakuta, Y.; Naito, T.; Takagawa, T.; Hanai, H.; Araki, H.; Sasaki, Y.; Sakuraba, H.; Sasaki, M.; Hisamatsu, T.; et al. Genetic Background of Mesalamine-induced Fever and Diarrhea in Japanese Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 28, 21–31. [Google Scholar] [CrossRef]
- Serna, M.J.; Rivera-Caravaca, J.M.; Gonzalez-Conejero, R.; Esteve-Pastor, M.A.; Valdes, M.; Vicente, V.; Lip, G.Y.H.; Roldan, V.; Marin, F. Pharmacogenetics of vitamin K antagonists and bleeding risk prediction in atrial fibrillation. Eur. J. Clin. Investig. 2018, 48, e12929. [Google Scholar] [CrossRef]
- Redensek, S.; Bizjan, B.J.; Trost, M.; Dolzan, V. Clinical and Clinical-Pharmacogenetic Models for Prediction of the Most Common Psychiatric Complications Due to Dopaminergic Treatment in Parkinson’s Disease. Int. J. Neuropsychopharmacol. 2020, 23, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Redensek, S.; Jenko Bizjan, B.; Trost, M.; Dolzan, V. Clinical-Pharmacogenetic Predictive Models for Time to Occurrence of Levodopa Related Motor Complications in Parkinson’s Disease. Front. Genet. 2019, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- Park, M.W.; Her, S.H.; Kim, C.J.; SunCho, J.; Park, G.M.; Kim, T.S.; Choi, Y.S.; Park, C.S.; Koh, Y.S.; Park, H.J.; et al. Evaluation of the incremental prognostic value of the combination of CYP2C19 poor metabolizer status and ABCB1 3435 TT polymorphism over conventional risk factors for cardiovascular events after drug-eluting stent implantation in East Asians. Genet. Med. 2016, 18, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Fotheringham, S.; Chegwidden, L.; Lucas, M.; Kerr, R.; Mozolowski, G.; Rosmarin, D.; Taylor, J.C.; Tomlinson, I.; Kerr, D. An Evaluation of the Diagnostic Accuracy of a Panel of Variants in DPYD and a Single Variant in ENOSF1 for Predicting Common Capecitabine Related Toxicities. Cancers 2021, 13, 1497. [Google Scholar] [CrossRef]
- Ooi, B.N.; Ying, A.F.; Koh, Y.Z.; Jin, Y.; Yee, S.W.; Lee, J.H.; Chong, S.S.; Tan, J.W.; Liu, J.; Lee, C.G.; et al. Robust Performance of Potentially Functional SNPs in Machine Learning Models for the Prediction of Atorvastatin-Induced Myalgia. Front. Pharmacol. 2021, 12, 605764. [Google Scholar] [CrossRef]
- Federico, N.; Stefania, F.F.; Rosalba, M.; Stefania, C.; Raffaella, G.; Giovanni, F.; Gabriele, I.; Antonia, M.; Carlotta, A.; Alfredo, F.; et al. Is a pharmacogenomic panel useful to estimate the risk of oxaliplatin-related neurotoxicity in colorectal cancer patients? Pharm. J. 2019, 19, 465–472. [Google Scholar] [CrossRef]
- Milosevic, G.; Kotur, N.; Krstovski, N.; Lazic, J.; Zukic, B.; Stankovic, B.; Janic, D.; Katsila, T.; Patrinos, G.P.; Pavlovic, S.; et al. Variants in TPMT, ITPA, ABCC4 and ABCB1 Genes as Predictors of 6-mercaptopurine Induced Toxicity in Children with Acute Lymphoblastic Leukemia. J. Med. Biochem. 2018, 37, 320–327. [Google Scholar] [CrossRef]
- Marcath, L.A.; Kidwell, K.M.; Vangipuram, K.; Gersch, C.L.; Rae, J.M.; Burness, M.L.; Griggs, J.J.; Van Poznak, C.; Hayes, D.F.; Smith, E.M.L.; et al. Genetic variation in EPHA contributes to sensitivity to paclitaxel-induced peripheral neuropathy. Br. J. Clin. Pharmacol. 2020, 86, 880–890. [Google Scholar] [CrossRef]
- Innocenti, F.; Kroetz, D.L.; Schuetz, E.; Dolan, M.E.; Ramirez, J.; Relling, M.; Chen, P.; Das, S.; Rosner, G.L.; Ratain, M.J. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J. Clin. Oncol. 2009, 27, 2604–2614. [Google Scholar] [CrossRef]
- Cummins, N.W.; Neuhaus, J.; Chu, H.; Neaton, J.; Wyen, C.; Rockstroh, J.K.; Skiest, D.J.; Boyd, M.A.; Khoo, S.; Rotger, M.; et al. Investigation of Efavirenz Discontinuation in Multi-ethnic Populations of HIV-positive Individuals by Genetic Analysis. EBioMedicine 2015, 2, 706–712. [Google Scholar] [CrossRef]
- Chen, S.; Villeneuve, L.; Jonker, D.; Couture, F.; Laverdiere, I.; Cecchin, E.; Innocenti, F.; Toffoli, G.; Levesque, E.; Guillemette, C. ABCC5 and ABCG1 polymorphisms predict irinotecan-induced severe toxicity in metastatic colorectal cancer patients. Pharm. Genom. 2015, 25, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, J.G.; Castagnino, J.P.; Aidar, O.; Musella, R.M.; Frias, A.; Visca, M.; Nogueras, M.; Costa, L.; Perez, A.; Caradonna, F.; et al. Effect of gene-gene and gene-environment interactions associated with antituberculosis drug-induced hepatotoxicity. Pharm. Genom. 2017, 27, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Biesiada, J.; Chidambaran, V.; Wagner, M.; Zhang, X.; Martin, L.J.; Meller, J.; Sadhasivam, S. Genetic risk signatures of opioid-induced respiratory depression following pediatric tonsillectomy. Pharmacogenomics 2014, 15, 1749–1762. Available online: http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=medp&NEWS=N&AN=25493568NS- (accessed on 28 November 2021). [CrossRef] [PubMed]
- Anandi, P.; Dickson, A.L.; Feng, Q.; Wei, W.Q.; Dupont, W.D.; Plummer, D.; Liu, G.; Octaria, R.; Barker, K.A.; Kawai, V.K.; et al. Combining clinical and candidate gene data into a risk score for azathioprine-associated leukopenia in routine clinical practice. Pharm. J. 2020, 20, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Abaji, R.; Ceppi, F.; Patel, S.; Gagne, V.; Xu, C.J.; Spinella, J.F.; Colombini, A.; Parasole, R.; Buldini, B.; Basso, G.; et al. Genetic risk factors for VIPN in childhood acute lymphoblastic leukemia patients identified using whole-exome sequencing. Pharmacogenomics 2018, 19, 1181–1193. [Google Scholar] [CrossRef]
- Mega, J.L.; Walker, J.R.; Ruff, C.T.; Vandell, A.G.; Nordio, F.; Deenadayalu, N.; Murphy, S.A.; Lee, J.; Mercuri, M.F.; Giugliano, R.P.; et al. Genetics and the clinical response to warfarin and edoxaban: Findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 2015, 385, 2280–2287. [Google Scholar] [CrossRef]
- Hamberg, A.K.; Friberg, L.E.; Hanseus, K.; Ekman-Joelsson, B.M.; Sunnegardh, J.; Jonzon, A.; Lundell, B.; Jonsson, E.N.; Wadelius, M. Warfarin dose prediction in children using pharmacometric bridging—Comparison with published pharmacogenetic dosing algorithms. Eur. J. Clin. Pharmacol. 2013, 69, 1275–1283. [Google Scholar] [CrossRef]
- Brugts, J.J.; Isaacs, A.; Boersma, E.; van Duijn, C.M.; Uitterlinden, A.G.; Remme, W.; Bertrand, M.; Ninomiya, T.; Ceconi, C.; Chalmers, J.; et al. Genetic determinants of treatment benefit of the angiotensin-convertingenzyme-inhibitor perindopril in patients with stable coronary arterydisease. Eur. Heart J. 2010, 31, 1854–1864. [Google Scholar] [CrossRef]
- Wu, X.; Lu, C.; Ye, Y.; Chang, J.; Yang, H.; Lin, J.; Gu, J.; Hong, W.K.; Stewart, D.; Spitz, M.R. Germline genetic variations in drug action pathways predict clinical outcomes in advanced lung cancer treated with platinum-based chemotherapy. Pharm. Genom. 2008, 18, 955–965. [Google Scholar] [CrossRef]
- Oemrawsingh, R.M.; Akkerhuis, K.M.; van Vark, L.C.; Ken Redekop, W.; Rudez, G.; Remme, W.J.; Bertrand, M.E.; Fox, K.M.; Ferrari, R.; Jan Danser, A.H.; et al. Individualized angiotensin-converting enzyme (ACE)-inhibitor therapy in stable coronary artery disease based on clinical and pharmacogenetic determinants: The PERindopril GENEtic (PERGENE) risk model. J. Am. Heart Assoc. 2015, 5, e002688. [Google Scholar] [CrossRef]
- Sensorn, I.; Sukasem, C.; Sirachainan, E.; Chamnanphon, M.; Pasomsub, E.; Trachu, N.; Supavilai, P.; Pinthong, D.; Wongwaisayawan, S. ABCB1 and ABCC2 and the risk of distant metastasis in Thai breast cancer patients treated with tamoxifen. OncoTargets Ther. 2016, 9, 2121–2129. [Google Scholar] [CrossRef][Green Version]
- Lorés-Motta, L.; van Asten, F.; Muether, P.S.; Smailhodzic, D.; Groenewoud, J.M.; Omar, A.; Chen, J.; Koenekoop, R.K.; Fauser, S.; Hoyng, C.B.; et al. A genetic variant in NRP1 is associated with worse response to ranibizumab treatment in neovascular age-related macular degeneration. Pharm. Genom. 2016, 26, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Gagno, S.; D’Andrea, M.R.; Mansutti, M.; Zanusso, C.; Puglisi, F.; Dreussi, E.; Montico, M.; Biason, P.; Cecchin, E.; Iacono, D.; et al. A New Genetic Risk Score to Predict the Outcome of Locally Advanced or Metastatic Breast Cancer Patients Treated with First-Line Exemestane: Results From a Prospective Study. Clin. Breast Cancer 2019, 19, 137–145.e4. [Google Scholar] [CrossRef] [PubMed]
- Gui, H.; Zeld, N.; Williams, L.K.; Lanfear, D.E.; Sabbah, H.N.; Li, J.; She, R.; Luzum, J.A.; Donahue, M.P.; Kraus, W.E.; et al. Polygenic Score for beta-Blocker Survival Benefit in European Ancestry Patients with Reduced Ejection Fraction Heart Failure. Circ. Heart Fail. 2020, 13, e007012. [Google Scholar] [CrossRef]
- Grželj, J.; Mlinarič-Raščan, I.; Marko, P.B.; Marovt, M.; Gmeiner, T.; Šmid, A. Polymorphisms in GNMT and DNMT3b are associated with methotrexate treatment outcome in plaque psoriasis. Biomed. Pharmacother. 2021, 138, 111456. [Google Scholar] [CrossRef]
- Duconge, J.; Santiago, E.; Hernandez-Suarez, D.F.; Moneró, M.; López-Reyes, A.; Rosario, M.; Renta, J.Y.; González, P.; Ileana Fernández-Morales, L.; Antonio Vélez-Figueroa, L.; et al. Pharmacogenomic polygenic risk score for clopidogrel responsiveness among Caribbean Hispanics: A candidate gene approach. Clin. Transl. Sci. 2021, 14, 2254–2266. [Google Scholar] [CrossRef]
- Yin, J.Y.; Li, X.; Li, X.P.; Xiao, L.; Zheng, W.; Chen, J.; Mao, C.X.; Fang, C.; Cui, J.J.; Guo, C.X.; et al. Prediction models for platinum-based chemotherapy response and toxicity in advanced NSCLC patients. Cancer Lett. 2016, 377, 65–73. [Google Scholar] [CrossRef]
- Wessels, J.A.M.; van der Kooij, S.M.; le Cessie, S.; Kievit, W.; Barerra, P.; Allaart, C.F.; Huizinga, T.W.J.; Guchelaar, H.J. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007, 56, 1765–1775. Available online: http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=med6&NEWS=N&AN=17530705 (accessed on 28 November 2021). [CrossRef]
- Wang, M.H.; Friton, J.J.; Raffals, L.E.; Leighton, J.A.; Pasha, S.F.; Picco, M.F.; Cushing, K.C.; Monroe, K.; Nix, B.D.; Newberry, R.D.; et al. Novel Genetic Risk Variants Can Predict Anti-TNF Agent Response in Patients With Inflammatory Bowel Disease. J. Crohn’s Colitis 2019, 13, 1036–1043. [Google Scholar] [CrossRef]
- Van der Leeuw, J.; Oemrawsingh, R.M.; van der Graaf, Y.; Brugts, J.J.; Deckers, J.W.; Bertrand, M.; Fox, K.; Ferrari, R.; Remme, W.J.; Simoons, M.L.; et al. Prediction of absolute risk reduction of cardiovascular events with perindopril for individual patients with stable coronary artery disease—Results from EUROPA. Int. J. Cardiol. 2015, 182, 194–199. [Google Scholar] [CrossRef]
- Sordillo, J.E.; Lutz, S.M.; McGeachie, M.J.; Lasky-Su, J.; Weiss, S.T.; Celedon, J.C.; Wu, A.C. Pharmacogenetic Polygenic Risk Score for Bronchodilator Response in Children and Adolescents with Asthma: Proof-of-Concept. J. Pers. Med. 2021, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.P.; Backman, J.D.; Reny, J.L.; Bergmeijer, T.O.; Mitchell, B.D.; Ritchie, M.D.; Dery, J.P.; Pakyz, R.E.; Gong, L.; Ryan, K.; et al. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel-treated patients. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 203–210, Erratum in Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 269. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32386422 (accessed on 28 November 2021). [CrossRef]
- Leusink, M.; Maitland-van der Zee, A.H.; Ding, B.; Drenos, F.; van Iperen, E.P.; Warren, H.R.; Caulfield, M.J.; Cupples, L.A.; Cushman, M.; Hingorani, A.D.; et al. A genetic risk score is associated with statin-induced low-density lipoprotein cholesterol lowering. Pharmacogenomics 2016, 17, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Hlavaty, T.; Ferrante, M.; Henckaerts, L.; Pierik, M.; Rutgeerts, P.; Vermeire, S. Predictive model for the outcome of infliximab therapy in Crohn’s disease based on apoptotic pharmacogenetic index and clinical predictors. Inflamm. Bowel Dis. 2007, 13, 372–379. Available online: http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=med6&NEWS=N&AN=17206723 (accessed on 28 November 2021). [CrossRef] [PubMed]
- Goricar, K.; Kovac, V.; Dolzan, V. Clinical-pharmacogenetic models for personalized cancer treatment: Application to malignant mesothelioma. Sci. Rep. 2017, 7, 46537. [Google Scholar] [CrossRef] [PubMed]
- Ciuculete, D.M.; Bandstein, M.; Benedict, C.; Waeber, G.; Vollenweider, P.; Lind, L.; Schioth, H.B.; Mwinyi, J. A genetic risk score is significantly associated with statin therapy response in the elderly population. Clin. Genet. 2017, 91, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Wong, H.S.C.; Chang, W.P.; Chen, B.K.; Wu, M.S.; Yang, K.D.; Hsieh, K.S.; Hsu, Y.W.; Liu, S.F.; Liu, X.; et al. Prediction for Intravenous Immunoglobulin Resistance by Using Weighted Genetic Risk Score Identified from Genome-Wide Association Study in Kawasaki Disease. Circ. Cardiovasc. Genet. 2017, 10, e001625. [Google Scholar] [CrossRef]
- Nelveg-Kristensen, K.E.; Busk Madsen, M.; Torp-Pedersen, C.; Kober, L.; Egfjord, M.; Berg Rasmussen, H.; Riis Hansen, P. Pharmacogenetic Risk Stratification in Angiotensin-Converting Enzyme Inhibitor-Treated Patients with Congestive Heart Failure: A Retrospective Cohort Study. PLoS ONE 2015, 10, e0144195. [Google Scholar] [CrossRef]
- Fransen, J.; Kooloos, W.M.; Wessels, J.A.M.; Huizinga, T.W.J.; Guchelaar, H.J.; van Riel, P.L.C.M.; Barrera, P. Clinical pharmacogenetic model to predict response of MTX monotherapy in patients with established rheumatoid arthritis after DMARD failure. Pharmacogenomics 2012, 13, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Eektimmerman, F.; Allaart, C.F.; Hazes, J.M.; Madhar, M.B.; den Broeder, A.A.; Fransen, J.; Swen, J.J.; Guchelaar, H.J. Validation of a clinical pharmacogenetic model to predict methotrexate nonresponse in rheumatoid arthritis patients. Pharmacogenomics 2019, 20, 85–93. [Google Scholar] [CrossRef]
- Amos, W.; Driscoll, E.; Hoffman, J.I. Candidate genes versus genome-wide associations: Which are better for detecting genetic susceptibility to infectious disease? Proc. R. Soc. B Biol. Sci. 2011, 278, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- McInnes, G.; Yee, S.W.; Pershad, Y.; Altman, R.B. Genomewide Association Studies in Pharmacogenomics. Clin. Pharmacol. Ther. 2021, 110, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, D.E.; Marsh, S.; McLeod, H.L. Caution with beta1-adrenergic receptor genotyping. Clin. Pharmacol. Ther. 2004, 76, 185–186. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, C.B.; Kroese, M.; Moorthie, S. Understanding polygenic models, their development and the potential application of polygenic scores in healthcare. J. Med. Genet. 2020, 57, 725–732. [Google Scholar] [CrossRef]
- Curtis, M.J.; Abernethy, D.R. Replication—Why we need to publish our findings. Pharmacol. Res. Perspect. 2015, 3, e00164. [Google Scholar] [CrossRef]
- Agnelli, G.; Buller, H.R.; Cohen, A.; Curto, M.; Gallus, A.S.; Johnson, M.; Masiukiewicz, U.; Pak, R.; Thompson, J.; Raskob, G.E.; et al. Oral apixaban for the treatment of acute venous thromboembolism. N. Engl. J. Med. 2013, 369, 25–26. [Google Scholar] [CrossRef]
- Kirley, K.; Qato, D.M.; Kornfield, R.; Stafford, R.S.; Caleb Alexander, G. National Trends in Oral Anticoagulant Use in the United States, 2007–2011. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 615–621. [Google Scholar] [CrossRef]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; Eriksson, H.; Baanstra, D.; Schnee, J.; Goldhaber, S.Z. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N. Engl. J. Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef]
- Budnitz, D.S.; Lovegrove, M.C.; Shehab, N.; Richards, C.L. Emergency hospitalizations for adverse drug events in older Americans. N. Engl. J. Med. 2011, 365, 2002–2012. [Google Scholar] [CrossRef]
- Johnson, J.A.; Cavallari, L.H. Warfarin Pharmacogenetics. Trends Cardiovasc. Med. 2015, 25, 33–41. [Google Scholar] [CrossRef]
- Daly, A.K. Pharmacogenomics of anticoagulants: Steps toward personal dosage. Genome Med. 2009, 1, 1–4. [Google Scholar] [CrossRef]
- Loebstein, R.; Dvoskin, I.; Halkin, H.; Vecsler, M.; Lubetsky, A.; Rechavi, G.; Amariglio, N.; Cohen, Y.; Ken-Dror, G.; Almog, S.; et al. A coding VKORC1 Asp36Tyr polymorphism predisposes to warfarin resistance. Blood 2007, 109, 2477–2480. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.M.; Stewart, K.; Dooley, M. The ten most common adverse drug reactions (ADRs) in oncology patients: Do they matter to you? Supportive Care Cancer 2004, 12, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Cancer in Children in Canada (0–14 Years)—Canada.ca. Available online: https://www.canada.ca/en/public-health/services/chronic-diseases/cancer/cancer-children-canada-0-14-years.html (accessed on 18 May 2022).
- Impicciatore, P.; Choonara, I.; Clarkson, A.; Provasi, D.; Pandolfini, C.; Bonati, M. Incidence of adverse drug reactions in paediatric in/out-patients: A systematic review and meta-analysis of prospective studies. Br. J. Clin. Pharmacol. 2001, 52, 77–83. [Google Scholar] [CrossRef]
- Linskey, D.W.; Linskey, D.C.; McLeod, H.L.; Luzum, J.A. The need to shift pharmacogenetic research from candidate gene to genome-wide association studies. Pharmacogenomics 2021, 22, 1143–1150. [Google Scholar] [CrossRef]
- Maranville, J.C.; Cox, N.J. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharm. J. 2016, 16, 388–392. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Beesley, L.J.; Vandehaar, P.; Peng, R.B.; Salvatore, M.; Zawistowski, M.; Taliun, S.A.G.; Das, S.; Lefaive, J.; Kaleba, E.O.; et al. Exploring various polygenic risk scores for skin cancer in the phenomes of the Michigan genomics initiative and the UK Biobank with a visual catalog: PRSWeb. PLoS Genet. 2019, 15, e1008202. [Google Scholar] [CrossRef]
- Wagner, M.W.; Namdar, K.; Biswas, A.; Monah, S.; Khalvati, F.; Ertl-Wagner, B.B. Radiomics, machine learning, and artificial intelligence—What the neuroradiologist needs to know. Neuroradiology 2021, 63, 1957–1967. [Google Scholar] [CrossRef]
- Ngiam, K.Y.; Khor, I.W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 2019, 20, e262–e273. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.; Telkar, N.; Reiker, T.; Walters, R.G.; Lin, K.; Eriksson, A.; Gurdasani, D.; Gilly, A.; Southam, L.; Tsafantakis, E.; et al. The transferability of lipid loci across African, Asian and European cohorts. Nat. Commun. 2019, 10, 4330. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.; Shen, H.; Gelaye, B.; Meijsen, J.; Ressler, K.; Feldman, M.; Peterson, R.; Domingue, B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019, 10, 3328. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, H.; Harpak, A.; Agarwal, I.; Conley, D.; Pritchard, J.K.; Przeworski, M. Variable prediction accuracy of polygenic scores within an ancestry group. ELife 2020, 9, e48376. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; McGregor, T.L. Pharmacogenetics in clinical pediatrics: Challenges and strategies. Pers. Med. 2013, 10, 661–671. [Google Scholar] [CrossRef]
- Brock, P.R.; Bellman, S.C.; Yeomans, E.C.; Pinkerton, C.R.; Pritchard, J. Cisplatin ototoxicity in children: A practical grading system. Med. Pediatr. Oncol. 1991, 19, 295–300. [Google Scholar] [CrossRef]
- Freyer, D.R.; Chen, L.; Krailo, M.D.; Knight, K.; Villaluna, D.; Bliss, B.; Pollock, B.H.; Ramdas, J.; Lange, B.; Van Hoff, D.; et al. Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 63–74. [Google Scholar] [CrossRef]
- Knight, K.R.G.; Kraemer, D.F.; Neuwelt, E.A. Ototoxicity in children receiving platinum chemotherapy: Underestimating a commonly occurring toxicity that may influence academic and social development. J. Clin. Oncol. 2005, 23, 8588–8596. [Google Scholar] [CrossRef]
- Drögemöller, B.I.; Wright, G.E.B.; Lo, C.; Le, T.; Brooks, B.; Bhavsar, A.P.; Rassekh, S.R.; Ross, C.J.D.; Carleton, B.C. Pharmacogenomics of Cisplatin-Induced Ototoxicity: Successes, Shortcomings, and Future Avenues of Research. Clin. Pharmacol. Ther. 2019, 106, 350–359. [Google Scholar] [CrossRef]
- King, K.A.; Brewer, C.C. Clinical trials, ototoxicity grading scales and the audiologist’s role in therapeutic decision making. Int. J. Audiol. 2018, 57 (Suppl. S4), S89–S98. [Google Scholar] [CrossRef]
- Schmidt, C.M.; Bartholomäus, E.; Deuster, D.; Heinecke, A.; Dinnesen, A.G. The “Muenster classification” of high frequency hearing loss following cisplatin chemotherapy. HNO 2007, 55, 299–306. [Google Scholar] [CrossRef]
- National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Common Terminology Criteria for Adverse Events (CTCAE) v5.0. Published Online 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf (accessed on 14 January 2022).
- Chang, K.W.; Chinosornvatana, N. Practical grading system for evaluating cisplatin ototoxicity in children. J. Clin. Oncol. 2010, 28, 1788–1795. [Google Scholar] [CrossRef]
- Clemens, E.; Brooks, B.; De Vries, A.C.H.; van Grotel, M.; van den Heuvel-Eibrink, M.M.; Carleton, B. A comparison of the Muenster, SIOP Boston, Brock, Chang and CTCAEv4.03 ototoxicity grading scales applied to 3799 audiograms of childhood cancer patients treated with platinum-based chemotherapy. PLoS ONE 2019, 14, e0210646. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, D.; Stride, E.; Vallis, K.; Gooding, M.J. Applications and limitations of machine learning in radiation oncology. Br. J. Radiol. 2019, 92, 20190001. [Google Scholar] [CrossRef] [PubMed]
- Phang-Lyn, S.; Llerena, V.A. Biochemistry, Biotransformation; StatPearls: Tampa, FL, USA, 2021. [Google Scholar]
- Joober, R.; Schmitz, N.; Annable, L.; Boksa, P. Publication bias: What are the challenges and can they be overcome? J. Psychiatry Neurosci. 2012, 37, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, A.; Kleinerman, E.S. Anthracycline-Induced Cardiotoxicity: Causes, Mechanisms, and Prevention. Adv. Exp. Med. Biol. 2020, 1257, 181–192. [Google Scholar] [CrossRef]
- Callejo, A.; Sedó-Cabezón, L.; Juan, I.D.; Llorens, J. Cisplatin-induced ototoxicity: Effects, mechanisms and protection strategies. Toxics 2015, 3, 268–293. [Google Scholar] [CrossRef]
- Triarico, S.; Romano, A.; Attinà, G.; Capozza, M.A.; Maurizi, P.; Mastrangelo, S.; Ruggiero, A. Vincristine-Induced Peripheral Neuropathy (VIPN) in Pediatric Tumors: Mechanisms, Risk Factors, Strategies of Prevention and Treatment. Int. J. Mol. Sci. 2021, 22, 4112. [Google Scholar] [CrossRef]
- Tellor, K.B.; Nguyen, S.N.; Bultas, A.C.; Armbruster, A.L.; Greenwald, N.A.; Yancey, A.M. Evaluation of the impact of body mass index on warfarin requirements in hospitalized patients. Ther. Adv. Cardiovasc. Dis. 2018, 12, 207–216. [Google Scholar] [CrossRef]
- Johnson, J.A. Pharmacogenetics in clinical practice: How far have we come and where are we going? Pharmacogenomics 2013, 14, 835–843. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Haidar, C.E.; Wilkinson, M.R.; Crews, K.R.; Baker, D.K.; Kornegay, N.M.; Yang, W.; Pui, C.H.; Reiss, U.M.; Gaur, A.H.; et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 45–55. [Google Scholar] [CrossRef]
- Danahey, K.; Borden, B.A.; Furner, B.; Yukman, P.; Hussain, S.; Saner, D.; Volchenboum, S.L.; Ratain, M.J.; O’Donnell, P.H. Simplifying the use of pharmacogenomics in clinical practice: Building the genomic prescribing system. J. Biomed. Inform. 2017, 75, 110–121. [Google Scholar] [CrossRef]
- Eadon, M.T.; Desta, Z.; Levy, K.D.; Decker, B.S.; Pierson, R.C.; Pratt, V.M.; Callaghan, J.T.; Rosenman, M.B.; Carpenter, J.S.; Holmes, A.M.; et al. Implementation of a pharmacogenomics consult service to support the INGENIOUS trial. Clin. Pharmacol. Ther. 2016, 100, 63–66. [Google Scholar] [CrossRef]
- Lip, G.Y.H.; Frison, L.; Halperin, J.L.; Lane, D.A. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: The HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile INR, elderly, drugs/alcohol concomitantly) score. J. Am. Coll. Cardiol. 2011, 57, 173–180. [Google Scholar] [CrossRef]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G.M. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): The TRIPOD statement. Ann. Intern. Med. 2015, 162, 55–63. [Google Scholar] [CrossRef]
- Ware, M.P.H.E.B.; Schmitz, L.L.; Faul, M.P.H.J.; Gard, A.M.; Mitchell, C.; Smith, M.P.H.J.A.; Zhao, W.; Weir, D.; Kardia, S.L. Heterogeneity in polygenic scores for common human traits. BioRxiv 2017, 106062. [Google Scholar] [CrossRef]
- Wand, H.; Lambert, S.A.; Tamburro, C.; Iacocca, M.A.; O’Sullivan, J.W.; Sillari, C.; Kullo, I.J.; Rowley, R.; Dron, J.S.; Brockman, D.; et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 2021, 591, 211–219. [Google Scholar] [CrossRef]
- Moons, K.G.M.; Kengne, A.P.; Woodward, M.; Royston, P.; Vergouwe, Y.; Altman, D.G.; Grobbee, D.E. Risk prediction models: I. Development, internal validation, and assessing the incremental value of a new (bio)marker. Heart 2012, 98, 683–690. [Google Scholar] [CrossRef]
- Moons, K.G.M.; Kengne, A.P.; Grobbee, D.E.; Royston, P.; Vergouwe, Y.; Altman, D.G.; Woodward, M. Risk prediction models: II. External validation, model updating, and impact assessment. Heart 2012, 98, 691–698. [Google Scholar] [CrossRef]
- Moons, K.G.M. Criteria for Scientific Evaluation of Novel Markers: A Perspective. Clin. Chem. 2010, 56, 537–541. [Google Scholar] [CrossRef][Green Version]
- Ivanov, J.; Tu, J.V.; Naylor, C.D. Ready-Made, Recalibrated, or Remodeled? Circulation 1999, 99, 2098–2104. [Google Scholar] [CrossRef]
- What Do We Mean by Validating a Prognostic Model?—Altman—2000—Statistics in Medicine—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1002/ (accessed on 10 May 2022).
- Steyerberg, E.W.; Borsboom, G.J.J.M.; van Houwelingen, H.C.; Eijkemans, M.J.C.; Habbema, J.D.F. Validation and updating of predictive logistic regression models: A study on sample size and shrinkage. Stat. Med. 2004, 23, 2567–2586. [Google Scholar] [CrossRef]
- Moons, K.G.M.; Altman, D.G.; Vergouwe, Y.; Royston, P. Prognosis and prognostic research: Application and impact of prognostic models in clinical practice. BMJ 2009, 338, 1487–1490. [Google Scholar] [CrossRef]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Kerminen, S.; Martin, A.R.; Koskela, J.; Ruotsalainen, S.E.; Havulinna, A.S.; Surakka, I.; Palotie, A.; Perola, M.; Salomaa, V.; Daly, M.J.; et al. Geographic Variation and Bias in the Polygenic Scores of Complex Diseases and Traits in Finland. Am. J. Hum. Genet. 2019, 104, 1169–1181. [Google Scholar] [CrossRef]
- Kim, M.S.; Patel, K.P.; Teng, A.K.; Berens, A.J.; Lachance, J. Genetic disease risks can be misestimated across global populations 06 Biological Sciences 0604 Genetics. Genome Biol. 2018, 19, 179. [Google Scholar] [CrossRef]
- Slunecka, J.L.; van der Zee, M.D.; Beck, J.J.; Johnson, B.N.; Finnicum, C.T.; Pool, R.; Hottenga, J.J.; de Geus, E.J.; Ehli, E.A. Implementation and implications for polygenic risk scores in healthcare. Hum. Genom. 2021, 15, 46. [Google Scholar] [CrossRef]
- Abdullah-Koolmees, H.; van Keulen, A.M.; Nijenhuis, M.; Deneer, V.H.M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front. Pharmacol. 2021, 11, 25. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).