
Treatment of Mantle Cell Lymphoma in the Frontline Setting: Are We Ready for a Risk-Adapted Approach?

Abstract
1. Introduction
2. Prognostic Factors Determined at Initial Diagnosis
2.1. Morphology
2.2. MIPI Score
2.3. Proliferation Index
2.4. MIPI-c Score
2.5. p53 Alterations
2.6. IGHV Mutations
2.7. Complex Karyotype
2.8. Gene Expression Profiling
2.9. Other Molecular Markers under Investigation
2.10. Imaging at Diagnosis
3. Prognostic Factors Determined Following Treatment
3.1. Imaging after Treatment
3.2. Minimal Residual Disease
3.3. Indolent MCL
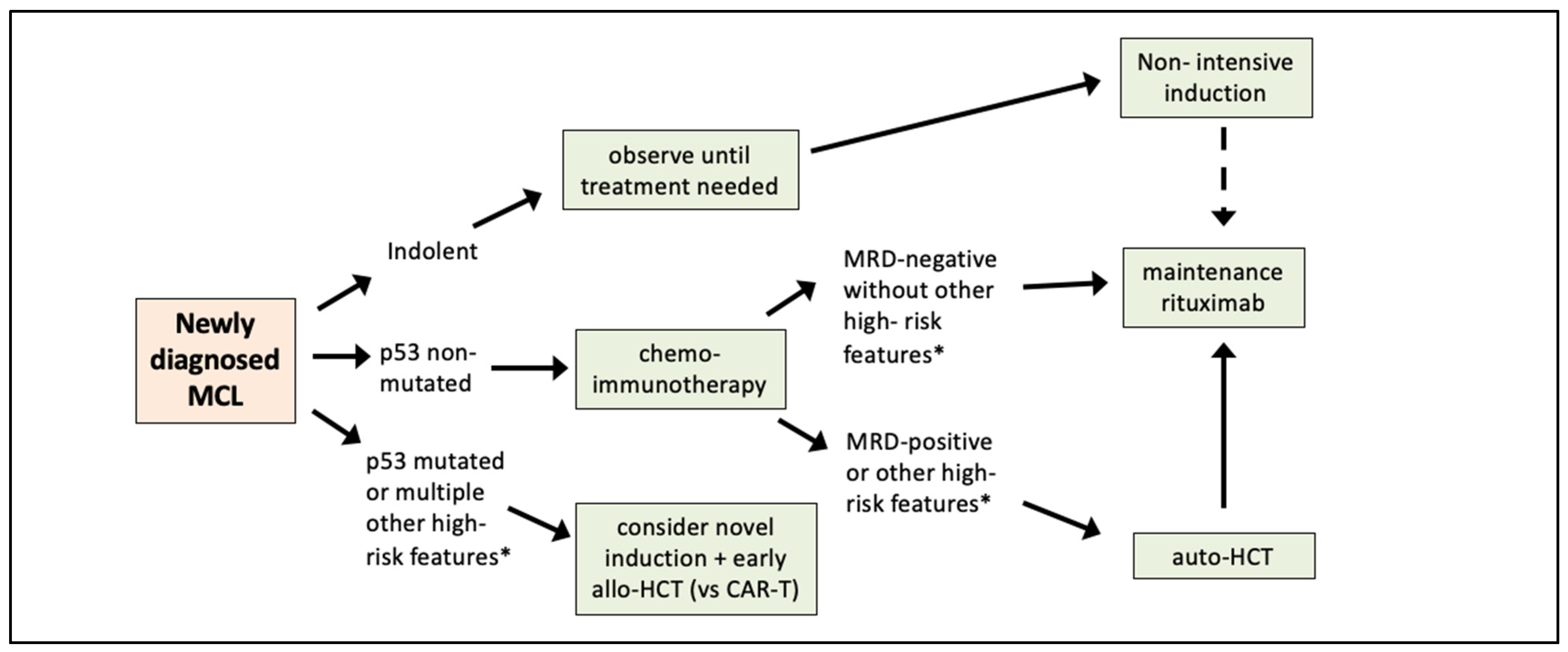
4. Moving from Prognostic Factors to Risk-Adapted Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Argatoff, L.H.; Connors, J.M.; Klasa, R.J.; Horsman, D.E.; Gascoyne, R.D. Mantle Cell Lymphoma: A Clinicopathologic Study of 80 Cases. Blood 1997, 89, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Samaha, H.D.C.; Ketterer, N.; Moullet, I.; Thieblemont, C.; Bouafia, F.; Callet-Bauchu, E.; Felman, P.; Berger, F.; Salles, G.; Coiffier, B. Mantle cell lymphoma: A retrospective study of 121 cases. Leukemia 1998, 12, 1281–1287. [Google Scholar] [CrossRef]
- Velders, G.A.; Kluin-Nelemans, J.; De Boer, C.J.; Hermans, J.; Noordijk, E.M.; Schuuring, E.; Kramer, M.H.; Van Deijk, W.A.; Rahder, J.B.; Kluin, P.M.; et al. Mantle-cell lymphoma: A population-based clinical study. J. Clin. Oncol. 1996, 14, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, S.; Bijnens, L.; Teodorovic, I.; Hagenbeek, A.; Meerwaldt, J.; Somers, R.; Thomas, J.; Noordijk, E.; De Wolf-Peeters, C. Clinical analysis of 670 cases in two trials of the European Organization for the Research and Treatment of Cancer Lymphoma Cooperative Group subtyped according to the Revised European-American Classification of Lymphoid Neoplasms: A comparison with the Working Formulation. Blood 1996, 87, 4358–4367. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.H.; Kolstad, A.; Laurell, A.; Jerkeman, M.; Räty, R.; Andersen, N.S.; Pedersen, L.B.; Eriksson, M.; Nordström, M.; Kimby, E.; et al. Nordic MCL2 trial update: Six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: Still very long survival but late relapses do occur. Br. J. Haematol. 2012, 158, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Hoster, E.; Walewski, J.; Bosly, A.; Stilgenbauer, S.; Thieblemont, C.; Szymczyk, M.; Bouabdallah, R.; Kneba, M.; Hallek, M.; et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): A randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016, 388, 565–575. [Google Scholar] [CrossRef]
- Romaguera, J.E.; Fayad, L.E.; Feng, L.; Hartig, K.; Weaver, P.; Rodriguez, M.A.; Hagemeister, F.B.; Pro, B.; McLaughlin, P.; Younes, A.; et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br. J. Haematol. 2010, 150, 200–208. [Google Scholar] [CrossRef]
- Riedell, P.A.; Hamadani, M.; Ahn, K.W.; Litovich, C.; Murthy, G.S.G.; Locke, F.L.; Brunstein, C.G.; Merryman, R.W.; Stiff, P.J.; Pawarode, A.; et al. Outcomes and Utilization Trends of Front-Line Autologous Hematopoietic Cell Transplantation for Man-tle Cell Lymphoma. Transplant. Cell. Ther. 2021, 27, 911.e1–911.e7. [Google Scholar] [CrossRef]
- Gerson, J.N.; Handorf, E.; Villa, D.; Gerrie, A.S.; Chapani, P.; Li, S.; Medeiros, L.J.; Wang, M.I.; Cohen, J.B.; Calzada, O.; et al. Survival Outcomes of Younger Patients With Mantle Cell Lymphoma Treated in the Rituximab Era. J. Clin. Oncol. 2019, 37, 471–480. [Google Scholar] [CrossRef]
- Jain, P.; Wang, M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am. J. Hematol. 2019, 94, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Eskelund, C.W.; Dimopoulos, K.; Kolstad, A.; Glimelius, I.; Räty, R.; Gjerdrum, L.M.R.; Sonnevi, K.; Josefsson, P.; Nilsson-Ehle, H.; Bentzen, H.H.N.; et al. Detailed Long-Term Follow-Up of Patients Who Relapsed After the Nordic Mantle Cell Lymphoma Trials: MCL2 and MCL3. HemaSphere 2020, 5, e510. [Google Scholar] [CrossRef]
- Tiemann, M.; Schrader, C.; Klapper, W.; Dreyling, M.H.; Campo, E.; Norton, A.; Berger, F.; Kluin, P.; Ott, G.; Pileri, S.; et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): A clinicopathological study from the European MCL Network. Br. J. Haematol. 2005, 131, 29–38. [Google Scholar] [CrossRef]
- Räty, R.F.K.; Joensuu, H.; Teerenhovi, L.; Elonen, E. Ki-67 expression level histological subtype and the International Prognostic. Eur. J. Haematol. 2002, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hoster, E.; Rosenwald, A.; Berger, F.; Bernd, H.-W.; Hartmann, S.; Loddenkemper, C.; Barth, T.F.; Brousse, N.; Pileri, S.; Rymkiewicz, G.; et al. Prognostic Value of Ki-67 Index, Cytology, and Growth Pattern in Mantle-Cell Lymphoma: Results From Randomized Trials of the European Mantle Cell Lymphoma Network. J. Clin. Oncol. 2016, 34, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Eskelund, C.W.; Kolstad, A.; Jerkeman, M.; Räty, R.; Laurell, A.; Eloranta, S.; Smedby, K.E.; Husby, S.; Pedersen, L.B.; Andersen, N.S.; et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): Prolonged remissions without survival plateau. Br. J. Haematol. 2016, 175, 410–418. [Google Scholar] [CrossRef] [PubMed]
- de Vos, S.K.U.; Hofmann, W.K.; Pinkus, G.S.; Swerdlow, S.H.; Wachsman, W.; Grogan, T.M.; Said, J.W.; Koeffler, H.P. Cell cycle altera-tions in the blastoid variant of mantle cell lymphoma (MCL-BV) as detected by gene expression profiling of mantle cell lym-phoma (MCL) and MCL-BV. Diagn. Mol. Pathol. 2003, 12, 35–43. [Google Scholar] [CrossRef]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, J.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef]
- Hoster, E.; Klapper, W.; Hermine, O.; Kluin-Nelemans, H.C.; Walewski, J.; van Hoof, A.; Trneny, M.; Geisler, C.H.; Di Raimondo, F.; Szymczyk, M.; et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J. Clin. Oncol. 2014, 32, 1338–1346. [Google Scholar] [CrossRef]
- Geisler, C.H.; Kolstad, A.; Laurell, A.; Räty, R.; Jerkeman, M.; Eriksson, M.; Nordström, M.; Kimby, E.; Boesen, A.M.; Nilsson-Ehle, H.; et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prog-nostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell trans-plantation (ASCT). Blood 2010, 115, 1530–1533. [Google Scholar] [CrossRef]
- Hsi, E.D.; Jung, S.-H.; Lai, R.; Johnson, J.L.; Cook, J.R.; Jones, D.; Devos, S.; Cheson, B.D.; Damon, L.E.; Said, J. Ki67 and PIM1 expression predict outcome in mantle cell lymphoma treated with high dose therapy, stem cell transplantation and rituximab: A Cancer and Leukemia Group B 59909 correlative science study. Leuk. Lymphoma 2008, 49, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, T.P.C.; Holler, S.; Mader, U.; Kalla, J.; Adam, P.; Ott, M.M.; Müller-Hermelink, H.K.; Rosenwald, A.; Ott, G. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood 2006, 107, 3407. [Google Scholar] [CrossRef] [PubMed]
- Eskelund, C.W.; Dahl, C.; Hansen, J.W.; Westman, M.; Kolstad, A.; Pedersen, L.B.; Montano-Almendras, C.P.; Husby, S.; Freiburghaus, C.; Ek, S.; et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood 2017, 130, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Delfau-Larue, M.-H.; Klapper, W.; Berger, F.; Jardin, F.; Briere, J.; Salles, G.; Casasnovas, R.-O.; Feugier, P.; Haioun, C.; Ribrag, V.; et al. High-dose cytarabine does not overcome the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood 2015, 126, 604–611. [Google Scholar] [CrossRef]
- Greiner, T.C.; Dasgupta, C.; Ho, V.V.; Weisenburger, D.D.; Smith, L.M.; Lynch, J.C.; Vose, J.M.; Fu, K.; Armitage, J.O.; Braziel, R.M.; et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2006, 103, 2352–2357. [Google Scholar] [CrossRef] [PubMed]
- Halldorsdottir, A.M.; Lundin, A.P.; Murray, F.; Mansouri, L.; Knuutila, S.; Sundstrom, C.; Laurell, A.; Ehrencrona, H.; Sander, B.; Rosenquist, R. Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia 2011, 25, 1904–1908. [Google Scholar] [CrossRef]
- Obr, A.; Procházka, V.; Jirkuvová, A.; Urbánková, H.; Kriegova, E.; Schneiderová, P.; Vatolíková, M.; Papajík, T. TP53 Mutation and Complex Karyotype Portends a Dismal Prognosis in Patients With Mantle Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 762–768. [Google Scholar] [CrossRef]
- Lin, R.J.; Ho, C.; Hilden, P.D.; Barker, J.N.; Giralt, S.A.; Hamlin, P.A.; Jakubowski, A.A.; Castro-Malaspina, H.R.; Robinson, K.S.; Papadopoulos, E.B.; et al. Allogeneic haematopoietic cell transplantation impacts on outcomes of mantle cell lymphoma with TP53 alterations. Br. J. Haematol. 2019, 184, 1006–1010. [Google Scholar]
- Thorselius, M.W.S.; Eriksson, I.; Thunberg, U.; Johnson, A.; Backlin, C.; Enblad, G.; Sundström, C.; Roos, G.; Rosenquist, R. Somatic hypermutation and V(H) gene usage in mantle cell lymphoma. Eur. J. Haematol. 2002, 68, 217–224. [Google Scholar] [CrossRef]
- Hadzidimitriou, A.; Agathangelidis, A.; Darzentas, N.; Murray, F.; Delfau-Larue, M.-H.; Pedersen, L.B.; Lopez, A.N.; Dagklis, A.; Rombout, P.; Beldjord, K.; et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood 2011, 118, 3088–3095. [Google Scholar] [CrossRef]
- Küppers, R.K.U.; Hansmann, M.L.; Rajewsky, K. Cellular morigin of human B-cell lymphomas. N. Engl. J. Med. 1999, 341, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Clot, G.; Royo, C.; Jares, P.; Hadzidimitriou, A.; Agathangelidis, A.; Bikos, V.; Darzentas, N.; Papadaki, T.; Salaverria, I.; et al. Molecular Subsets of Mantle Cell Lymphoma Defined by the IGHV Mutational Status and SOX11 Expression Have Distinct Biologic and Clinical Features. Cancer Res. 2012, 72, 5307–5316. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Terré, C.; Jardin, F.; Radford, I.; Roche-Lestienne, C.; Penther, D.; Bastard, C.; Rigaudeau, S.; Pilorge, S.; Morschhauser, F.; et al. Complex karyotype in mantle cell lymphoma is a strong prognostic factor for the time to treatment and overall survival, independent of the MCL international prognostic index. Genes Chromosom. Cancer 2013, 53, 106–116. [Google Scholar] [CrossRef]
- Greenwell, I.B.; Staton, A.D.; Lee, M.J.; Switchenko, J.M.; Saxe, D.F.; Maly, J.J.; Blum, K.A.; Grover, N.S.; Mathews, S.P.; Gordon, M.J.; et al. Complex karyotype in patients with mantle cell lymphoma predicts inferior survival and poor response to intensive induction therapy. Cancer 2018, 124, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.W.G.; Wiestner, A.; Chan, W.C.; Connors, J.M.; Campo, E.; Gascoyne, R.D.; Grogan, T.M.; Muller-Hermelink, H.K.; Smeland, E.B.; Chiorazzi, M.; et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003, 3, 185–197. [Google Scholar] [CrossRef]
- Scott, D.W.; Abrisqueta, P.; Wright, G.W.; Slack, G.W.; Mottok, A.; Villa, D.; Jares, P.; Rauert-Wunderlich, H.; Royo, C.; Clot, G.; et al. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J. Clin. Oncol. 2017, 35, 1668–1677. [Google Scholar] [CrossRef]
- Rauert-Wunderlich, H.; Mottok, A.; Scott, D.W.; Rimsza, L.M.; Ott, G.; Klapper, W.; Unterhalt, M.; Kluin-Nelemans, H.C.; Hermine, O.; Hartmann, S.; et al. Validation of the MCL35 gene expression proliferation assay in randomized trials of the Euro-pean Mantle Cell Lymphoma Network. Br. J. Haematol. 2019, 184, 616–624. [Google Scholar] [CrossRef]
- Ferrero, S.; Rossi, D.; Rinaldi, A.; Bruscaggin, A.; Spina, V.; Eskelund, C.W.; Evangelista, A.; Moia, R.; Kwee, I.; Dahl, C.; et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: A FIL study. Haematologica 2019, 105, 1604–1612. [Google Scholar] [CrossRef]
- Condoluci, A.; Rossi, D.; Zucca, E.; Cavalli, F. Toward a Risk-Tailored Therapeutic Policy in Mantle Cell Lymphoma. Curr. Oncol. Rep. 2018, 20, 79. [Google Scholar] [CrossRef]
- Jain, P.; Zhang, S.; Kanagal-Shamanna, R.; Ok, C.Y.; Nomie, K.; Gonzalez, G.N.; Gonzalez-Pagan, O.; Hill, H.A.; Lee, H.J.; Fayad, L.; et al. Genomic profiles and clinical outcomes of de novo blastoid/pleomorphic MCL are distinct from those of trans-formed MCL. Blood Adv. 2020, 4, 1038–1050. [Google Scholar] [CrossRef]
- Choe, J.Y.; Yun, J.Y.; Na, H.Y.; Huh, J.; Shin, S.J.; Kim, H.J.; Paik, J.H.; Kim, Y.A.; Nam, S.J.; Jeon, Y.K.; et al. MYC overexpression correlates with MYC amplification or translocation, and is associated with poor prognosis in mantle cell lymphoma. Histopathology 2016, 68, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Mas, R.M.V.; Navarro, A.; Salaverria, I.; Martín-Garcia, D.; Jares, P.; Giné, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef]
- Mohanty, A.S.N.; Das, M.; Pillai, R.; Chen, L.; Chen, R.W.; Amin, H.M.; Wang, M.; Marcucci, G.; Weisenburger, D.D.; Rosen, S.T.; et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget 2016, 7, 73558–73572. [Google Scholar] [CrossRef]
- Husby, S.; Ralfkiaer, U.; Garde, C.; Zandi, R.; Ek, S.; Kolstad, A.; Jerkeman, M.; Laurell, A.; Räty, R.; Pedersen, L.B.; et al. miR-18b overexpression identifies mantle cell lymphoma patients with poor outcome and improves the MI-PI-B prognosticator. Blood 2015, 125, 2669–2677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolstad, A.; Laurell, A.; Jerkeman, M.; Grønbæk, K.; Elonen, E.; Räty, R.; Pedersen, L.B.; Loft, A.; Bogsrud, T.V.; Kimby, E.; et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014, 123, 2953–2959. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Carlier, T.; Berriolo-Riedinger, A.; Casasnovas, O.; Gyan, E.; Meignan, M.; Moreau, A.; Burroni, B.; Djaileb, L.; Gressin, R.; et al. Prognostic value of FDG-PET in patients with mantle cell lymphoma: Results from the LyMa-PET Project. Haematologica 2020, 105, e33–e36. [Google Scholar] [CrossRef] [PubMed]
- Karam, M.; Ata, A.; Irish, K.; Feustel, P.J.; Mottaghy, F.M.; Stroobants, S.G.; Verhoef, G.E.; Chundru, S.; Douglas-Nikitin, V.; Wong, C.Y.O.; et al. FDG positron emission tomography/computed tomography scan may identify mantle cell lymphoma patients with unusually favorable outcome. Nucl. Med. Commun. 2009, 30, 770–778. [Google Scholar] [CrossRef]
- Bodet-Milin, C.; Touzeau, C.; Leux, C.; Sahin, M.; Moreau, A.; Maisonneuve, H.; Morineau, N.; Jardel, H.; Moreau, P.; Gallazini-Crépin, C.; et al. Prognostic impact of 18F-fluoro-deoxyglucose positron emission tomography in untreated mantle cell lymphoma: A retrospective study from the GOELAMS group. Eur. J. Pediatr. 2010, 37, 1633–1642. [Google Scholar] [CrossRef]
- Kedmi, M.; Avivi, I.; Ribakovsky, E.; Benyamini, N.; Davidson, T.; Goshen, E.; Tadmor, T.; Nagler, A.; Avigdor, A. Is there a role for therapy response assessment with 2-[fluorine-18] fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography in mantle cell lymphoma? Leuk Lymphoma 2014, 55, 2484–2489. [Google Scholar] [CrossRef]
- Hosein, P.J.; Pastorini, V.H.; Paes, F.M.; Eber, D.; Chapman, J.R.; Serafini, A.N.; Alizadeh, A.A.; Lossos, I.S. Utility of positron emission tomography scans in mantle cell lymphoma. Am. J. Hematol. 2011, 86, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Albano, D.; Bosio, G.; Bianchetti, N.; Pagani, C.; Re, A.; Tucci, A.; Giubbini, R.; Bertagna, F. Prognostic role of baseline 18F-FDG PET/CT metabolic parameters in mantle cell lymphoma. Ann. Nucl. Med. 2019, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Noring, K.; Carlsten, M.; Sonnevi, K.; Wahlin, B.E. The value of complete remission according to positron emission tomography prior to autologous stem cell transplantation in lymphoma: A population-based study showing improved outcome. BMC Cancer 2021, 21, 500. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Svoboda, J.; Feldman, T.; Zielonka, T.; Agress, H.; Panush, D.; Miller, M.; Toth, P.; Lizotte, P.M.; Nasta, S.; et al. Post-treatment (not interim) positron emission tomography-computed tomography scan status is highly predictive of outcome in mantle cell lymphoma patients treated with R-HyperCVAD. Cancer 2012, 118, 3565–3570. [Google Scholar] [CrossRef] [PubMed]
- Karls, S.; Shah, H.; Jacene, H. PET/CT for Lymphoma Post-therapy Response Assessment in Other Lymphomas, Response Assessment for Autologous Stem Cell Transplant, and Lymphoma Follow-up. Semin. Nucl. Med. 2018, 48, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, S.; Ritgen, M.; Kneba, M. Flow cytometric MRD detection in selected mature B-cell malignancies. In Lymphoma: Methods and Protocols; Küppers, R., Ed.; Humana Press: Essen, Germany, 2019; pp. 149–174. [Google Scholar]
- Scheijen, B.; Meijers, R.W.; Rijntjes, J.; van der Klift, M.Y.; Möbs, M.; Steinhilber, J.; Reigl, T.; van den Brand, M.; Kotrová, M.; Ritter, J.M.; et al. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: A technical feasibility study by EuroClonality-NGS. Leukemia 2019, 33, 2227–2240. [Google Scholar] [CrossRef]
- Sriram, D.; Lakhotia, R.; Fenske, T.S. Measurement of circulating tumor DNA to guide management of patients with lymphoma. Measurement 2019, 17, 509–517. [Google Scholar]
- Liu, H.; Johnson, J.L.; Koval, G.; Malnassy, G.; Sher, D.; Damon, L.E.; Hsi, E.D.; Bucci, D.M.; Linker, C.A.; Cheson, B.D.; et al. Detection of minimal residual disease following induction immunochemotherapy predicts progression free survival in mantle cell lymphoma: Final results of CALGB 59909. Haematologica 2012, 97, 579–585. [Google Scholar] [CrossRef]
- Kluin-Nelemans, H.C.; Hoster, E.; Hermine, O.; Walewski, J.; Geisler, C.H.; Trneny, M.; Stilgenbauer, S.; Kaiser, F.; Doorduijn, J.K.; Salles, G.; et al. Treatment of Older Patients With Mantle Cell Lymphoma (MCL): Long-Term Follow-Up of the Randomized European MCL Elderly Trial. J. Clin. Oncol. 2020, 38, 248–256. [Google Scholar] [CrossRef]
- Pott, C.; Hoster, E.; Delfau-Larue, M.-H.; Beldjord, K.; Böttcher, S.; Asnafi, V.; Plonquet, A.; Siebert, R.; Callet-Bauchu, E.; Andersen, N.; et al. Molecular remission is an independent predictor of clinical outcome in patients with mantle cell lymphoma after combined immunochemotherapy: A European MCL intergroup study. Blood 2010, 115, 3215–3223. [Google Scholar] [CrossRef]
- Cowan, A.J.; Stevenson, P.A.; Cassaday, R.D.; Graf, S.A.; Fromm, J.R.; Wu, D.; Holmberg, L.A.; Till, B.G.; Chauncey, T.R.; Smith, S.D.; et al. Pretransplantation Minimal Residual Disease Predicts Survival in Patients with Mantle Cell Lymphoma Undergoing Autologous Stem Cell Transplantation in Complete Remission. Biol. Blood Marrow Transplant. 2015, 22, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Gressin, R.; Daguindau, N.; Tempescul, A.; Moreau, A.; Carras, S.; Tchernonog, E.; Schmitt, A.; Houot, R.; Dartigeas, C.; Pignon, J.M.; et al. A phase 2 study of rituximab, bendamustine, bortezomib and dexamethasone for first-line treatment of older patients with mantle cell lymphoma. Haematologica 2018, 104, 138–146. [Google Scholar] [CrossRef]
- Klener, P.; Fronkova, E.; Kalinova, M.; Belada, D.; Forsterova, K.; Pytlik, R.; Blahovcova, P.; Simkovic, M.; Salek, D.; Mocikova, H.; et al. Potential loss of prognostic significance of minimal residual disease assessment after R-CHOP-based induction in elderly patients with mantle cell lymphoma in the era of rituximab maintenance. Hematol. Oncol. 2018, 36, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Jegede, O.; Parekh, S.; Hanson, C.A.; Martin, P.; Till, B.G.; Crump, M.; Leonard, J.P.; Friedberg, J.W.; Kahl, B.S.; et al. Minimal Residual Disease (MRD) Assessment in the ECOG1411 Randomized Phase 2 Trial of Front-Line Bendamustine-Rituximab (BR)-Based Induction Followed By Rituximab (R) ± Lenalidomide (L) Consolidation for Mantle Cell Lymphoma (MCL). Blood 2019, 134, 751. [Google Scholar] [CrossRef]
- Merryman, R.W.; Edwin, N.; Redd, R.; Bsat, J.; Chase, M.; LaCasce, A.; Freedman, A.; Jacobson, C.; Fisher, D.; Ng, S.; et al. Rituximab/bendamustine and rituximab/cytarabine induction therapy for transplant-eligible mantle cell lymphoma. Blood Adv. 2020, 4, 858–867. [Google Scholar] [CrossRef]
- Andersen, N.S.; Pedersen, L.B.; Laurell, A.; Elonen, E.; Kolstad, A.; Boesen, A.M.; Pedersen, L.M.; Lauritzsen, G.F.; Ekanger, R.; Nilsson-Ehle, H.; et al. Pre-emptive treatment with rituximab of molecular relapse after autologous stem cell transplantation in mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 4365–4370. [Google Scholar] [CrossRef]
- Wu, X.; Lu, H.; Pang, T.; Li, X.; Luo, H.; Tan, H.; Liu, S. Association of minimal residual disease levels with clinical outcomes in patients with mantle cell lymphoma: A meta-analysis. Leuk. Res. 2021, 108, 106605. [Google Scholar] [CrossRef]
- Orchard, J.; Garand, R.; Davis, Z.; Babbage, G.; Sahota, S.; Matutes, E.; Catovsky, D.; Thomas, P.W.; Avet-Loiseau, H.; Oscier, D. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood 2003, 101, 4975–4981. [Google Scholar] [CrossRef]
- Martin, P.; Chadburn, A.; Christos, P.; Weil, K.; Furman, R.R.; Ruan, J.; Elstrom, R.; Niesvizky, R.; Ely, S.; DiLiberto, M.; et al. Outcome of Deferred Initial Therapy in Mantle-Cell Lymphoma. J. Clin. Oncol. 2009, 27, 1209–1213. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Goyal, S.; Switchenko, J.; Calzada, O.; Churnetski, M.C.; Kolla, B.; Bachanova, V.; Gerson, J.N.; Barta, S.K.; Gordon, M.J.; et al. Intensive induction regimens after deferring initial therapy for mantle cell lymphoma are not associated with improved survival. Eur. J. Haematol. 2021, 107, 301–310. [Google Scholar] [CrossRef]
- Calzada, O.; Switchenko, J.M.; Maly, J.J.; Blum, K.A.; Grover, N.; Mathews, S.; Park, S.I.; Gordon, M.; Danilov, A.; Epperla, N.; et al. Deferred treatment is a safe and viable option for selected patients with mantle cell lymphoma. Leuk. Lymphoma 2018, 59, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Anderson, M.A.; Pott, C.; Agarwal, R.; Handunnetti, S.; Hicks, R.J.; Burbury, K.; Turner, G.; Di Iulio, J.; Bressel, M.; et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1211–1223. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Poor Prognostic Factor | References | Outcomes (p-Value) | ||
|---|---|---|---|---|
| PFS or TTF | OS | |||
| Morphology | Blastoid histology * | [2] | - | <0.001 |
| [12] | - | <0.0001 | ||
| [13] | - | NS | ||
| [14] | - | <0.001 | ||
| [15] | NS | NS | ||
| [16] | NS | NS | ||
| MIPI Score | Intermediate- and high-risk score versus low-risk score | [18] | - | <0.001 |
| [19] | <0.001 | <0.001 | ||
| [20] | - | <0.0001 | ||
| Proliferation Index | Ki-67 or MIB-1 | [14] | <0.001 | <0.001 |
| [21] | ≤ 0.030 | - | ||
| [22] | - | <0.001 | ||
| MIPI-c Score | Intermediate and high-risk | [15] | <0.001 | <0.001 |
| p53 Alterations | p53 deletions | [24] | - | 0.0003 |
| [25] | - | 0.081 | ||
| [26] | - | 0.7 | ||
| [23] | 0.003 | 0.002 | ||
| [38] | <0.0001 | <0.0001 | ||
| p53 mutations | [27] | <0.001 | <0.001 | |
| [23] | <0.0001 | <0.0001 | ||
| [25] | - | 0.0033 | ||
| [26] | - | 0.0006 | ||
| [38] | <0.001 | <0.001 | ||
| IGHV Mutations | IGHV unmutated status | [29] | - | NS |
| [32] | - | 0.004 | ||
| Complex Karyotype | ≥3 karyotypic abnormalities | [33] | - | 0.017 |
| [34] | <0.01 | <0.01 | ||
| [27] | 0.02 | 0.001 | ||
| Gene Expression Profiling | MCL-35 assay | [36] | - | <0.01 |
| [37] | <0.0001 | <0.0001 | ||
| Other Molecular Markers | KMT2D mutation | [38] | <0.001 | 0.002 |
| MIPI-g | [38] | <0.0001 | <0.0001 | |
| CDKN2A mutation | [24] | - | 0.0001 | |
| CDKN2A mutation + TP53 mutation | [24] | - | <0.0001 | |
| MYC over-expression (≥20%) | [40] | 0.001 | 0.002 | |
| NOTCH1 mutation | [42] | NS | 0.002 | |
| [43] | - | 0.026 | ||
| NOTCH1 + NOTCH2 mutations | [43] | - | 0.00034 | |
| NOTCH2 mutation | [43] | - | 0.00025 | |
| microRNA 18b + MIPI-c | [45] | <0.001 | <0.001 | |
| 18FDG-PET | SUVmax > 10.3 | [47] | <0.001 | <0.001 |
| SUVmax > 5 | [48] | <0.001 | <0.01 | |
| SUVmax > 6 | [49] | - | NS | |
| SUVmax > 10.3 + high MIPI | [47] | 0.0027 | 0.0002 | |
| Poor Prognostic Factor | References | Outcomes (p-Value) | ||
|---|---|---|---|---|
| PFS or TTF | OS | |||
| 18FDG-PET | Mid-induction therapy | [50] | NS | NS |
| [51] | NS | NS | ||
| [54] | NS | NS | ||
| Post-induction/before transplant | [46] | 0.017 | < 0.0001 | |
| [52] | < 0.001 | NS | ||
| [53] | 0.03 | 0.042 | ||
| [54] | 0.001 | NS | ||
| [50] | NS | NS | ||
| [51] | - | NS | ||
| Post-induction/before transplant SUVmax >5 | [48] | - | 0.01 | |
| Post-induction/post-auto-HCT | [50] | NS | NS | |
| [47] | 0.0209 | NS | ||
| Minimal Residual Disease | Post-induction/pre-auto-HCT | [59] | 0.016 | NS |
| [61] | 0.022 | NS | ||
| [61] | 0.021 | NS | ||
| [62] | 0.009 | 0.002 | ||
| [63] | < 0.0001 | < 0.0001 | ||
| [46] | 0.029 | NS | ||
| [68] | [HR 0.9, CI 0.84–0.97] | [HR 0.47, CI 0.31–0.72] | ||
| Post-induction/on maintenance therapy | [64] | NS | NS | |
| [65] | 0.002 | - | ||
| Post-induction/post-auto-HCT | [46] | 0.0001 | - | |
| [68] | [HR 0.11, CI 0.05–0.27] | [HR 0.14, CI 0.06–0.33] | ||
| Indolent MCL | Non-nodal disease vs. nodal disease | [69] | - | 0.005 |
| Delayed treatment ≥ 3months | [12] | < 0.0001 | < 0.0001 | |
| [70] | - | 0.0038 | ||
| [71] | - | 0.03 | ||
| [72] | NS | NS | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammons, L.; Fenske, T.S. Treatment of Mantle Cell Lymphoma in the Frontline Setting: Are We Ready for a Risk-Adapted Approach? J. Pers. Med. 2022, 12, 1134. https://doi.org/10.3390/jpm12071134
Hammons L, Fenske TS. Treatment of Mantle Cell Lymphoma in the Frontline Setting: Are We Ready for a Risk-Adapted Approach? Journal of Personalized Medicine. 2022; 12(7):1134. https://doi.org/10.3390/jpm12071134
Chicago/Turabian StyleHammons, Lindsay, and Timothy S. Fenske. 2022. "Treatment of Mantle Cell Lymphoma in the Frontline Setting: Are We Ready for a Risk-Adapted Approach?" Journal of Personalized Medicine 12, no. 7: 1134. https://doi.org/10.3390/jpm12071134
APA StyleHammons, L., & Fenske, T. S. (2022). Treatment of Mantle Cell Lymphoma in the Frontline Setting: Are We Ready for a Risk-Adapted Approach? Journal of Personalized Medicine, 12(7), 1134. https://doi.org/10.3390/jpm12071134