
Modeling a Novel Variant of Glycogenosis IXa Using a Clonal Inducible Reprogramming System to Generate “Diseased” Hepatocytes for Accurate Diagnosis

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Lentivirus Generation
2.2. Cell Culture and Imaging
2.3. Reprogramming into Hepatocyte-Like Cells
2.4. Sequencing and RT-qPCR
2.5. Immunofluorescence, PAS Staining, Glycogen Content, and Immunoblotting
3. Results
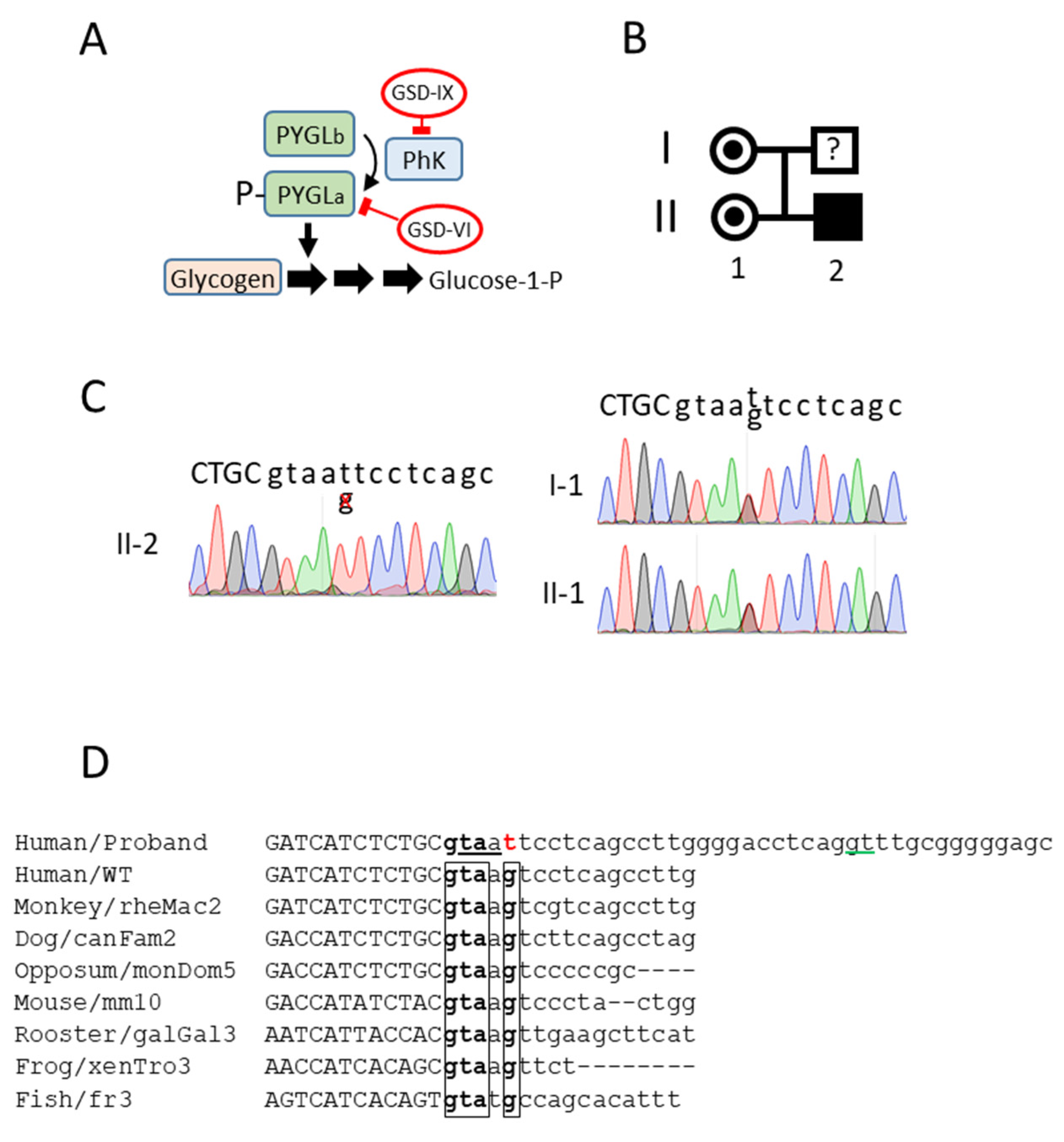
3.1. A Novel Mutation in the Donor Splice Site in Intron 23 of the PHKA2 Gene
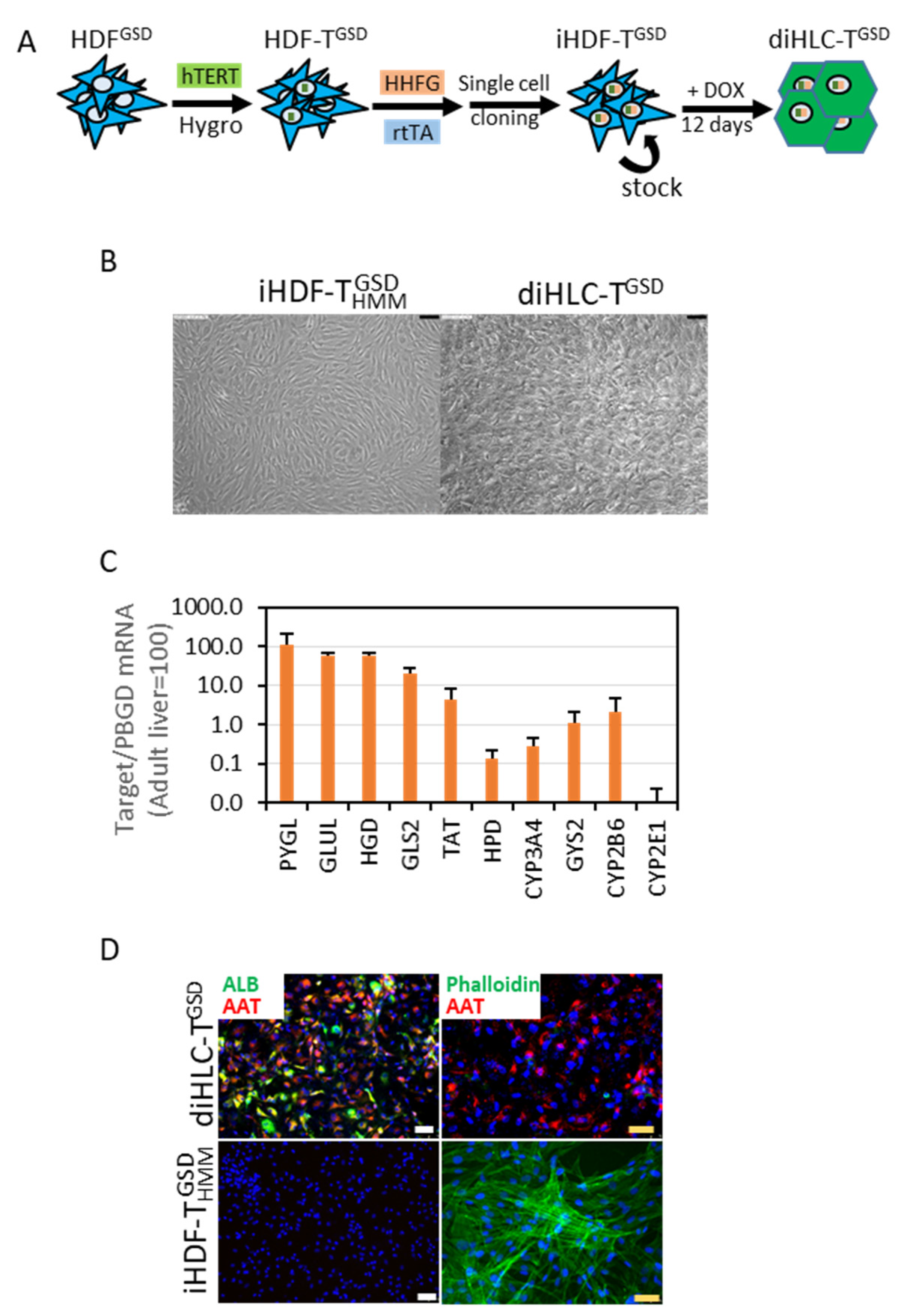
3.2. Implementing a Robust and Simple Method to Generate Hepatocytes from Patient-Specific Dermal Fibroblasts
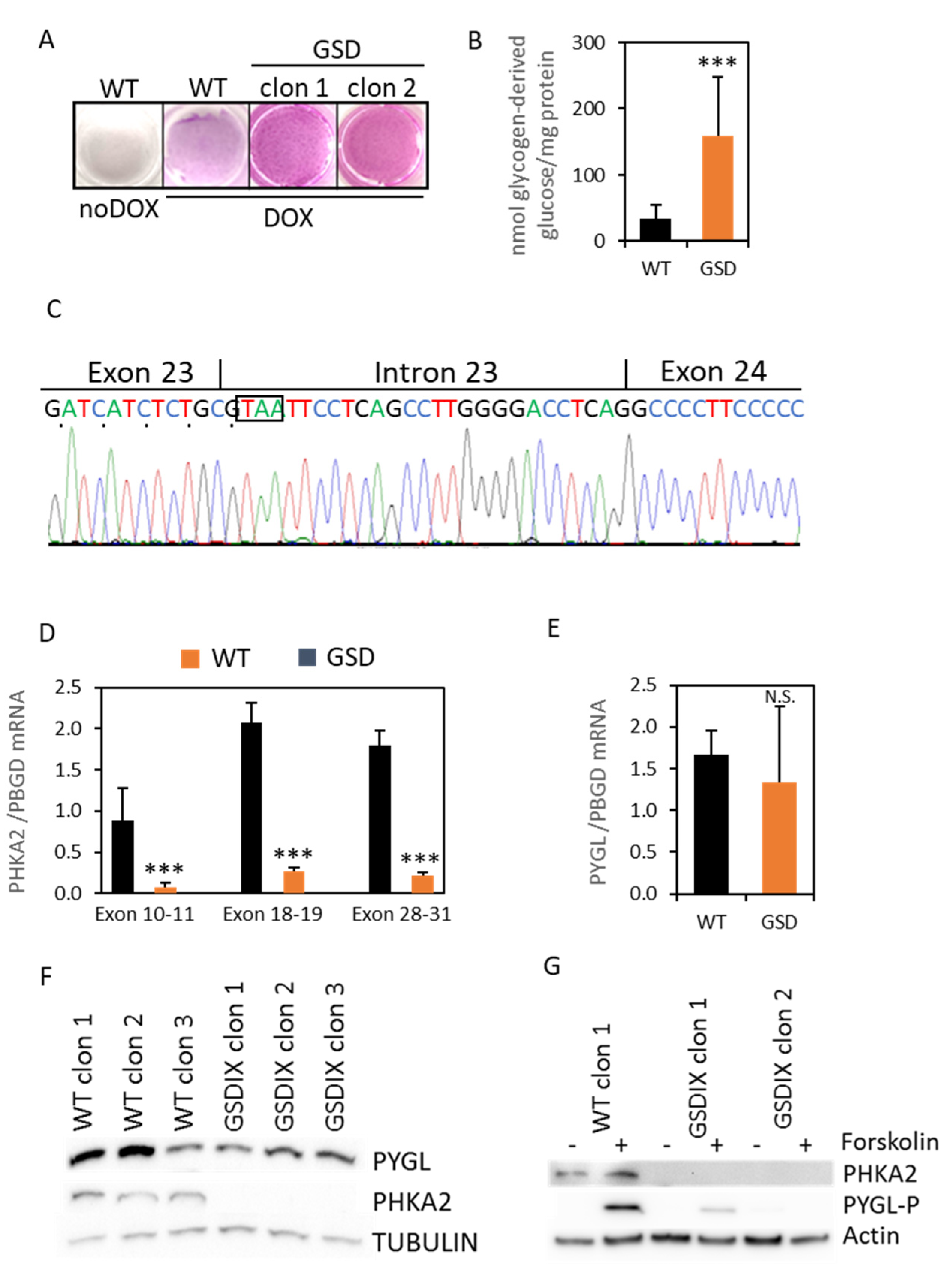
3.3. diHLC-TGSD Display Characteristic Features of GSD Type IX Diseased Hepatocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The Incidence of Inherited Metabolic Disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscher, A.; Patel, J.; Hewson, S.; Nagy, L.; Feigenbaum, A.; Kronick, J.; Raiman, J.; Schulze, A.; Siriwardena, K.; Mercimek-Mahmutoglu, S. The Natural History of Glycogen Storage Disease Types VI and IX: Long-Term Outcome from the Largest Metabolic Center in Canada. Mol. Genet. Metab. 2014, 113, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.; Novoa, W.B.; Krebs, E.G.; Fischer, E.H. Further Studies on the Site Phosphorylated in the Phosphorylase B to A Reaction. Biochemistry 1964, 3, 542–551. [Google Scholar] [CrossRef]
- Hendrickx, J.; Lee, P.; Keating, J.P.; Carton, D.; Sardharwalla, I.B.; Tuchman, M.; Baussan, C.; Willems, P.J. Complete Genomic Structure and Mutational Spectrum of PHKA2 in Patients with X-Linked Liver Glycogenosis Type I and II. Am. J. Hum. Genet. 1999, 64, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Heller, S.; Worona, L.; Consuelo, A. Nutritional Therapy for Glycogen Storage Diseases. J. Pediatr. Gastroenterol. Nutr. 2008, 47, S15. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Llorens, G.; Martinez-Sena, T.; Pareja, E.; Castell, J.V.; Bort, R. A Robust Reprogramming Strategy for Generating Hepatocyte-like Cells Usable in Pharmaco-Toxicological Studies. Stem Cell Res. Ther. 2022, Submitted. [Google Scholar]
- Hayer, A.; Shao, L.; Chung, M.; Joubert, L.-M.; Yang, H.W.; Tsai, F.-C.; Bisaria, A.; Betzig, E.; Meyer, T. Engulfed Cadherin Fingers Are Polarized Junctional Structures between Collectively Migrating Endothelial Cells. Nat. Cell Biol. 2016, 18, 1311–1323. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Cook, E.G.; Gao, Q.; Mitalipova, M.; Jaenisch, R. A Drug-Inducible System for Direct Reprogramming of Human Somatic Cells to Pluripotency. Cell Stem Cell 2008, 3, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Ballester, M.; Bolonio, M.; Santamaria, R.; Castell, J.V.; Ribes-Koninckx, C.; Bort, R. Direct Conversion of Human Fibroblast to Hepatocytes Using a Single Inducible Polycistronic Vector. Stem Cell Res. Ther. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Falanga, V.; Kirsner, R.S. Low Oxygen Stimulates Proliferation of Fibroblasts Seeded as Single Cells. J. Cell. Physiol. 1993, 154, 506–510. [Google Scholar] [CrossRef]
- Serrano, F.; García-Bravo, M.; Blazquez, M.; Torres, J.; Castell, J.V.; Segovia, J.C.; Bort, R. Silencing of Hepatic Fate-Conversion Factors Induce Tumorigenesis in Reprogrammed Hepatic Progenitor-like Cells. Stem Cell Res. Ther. 2016, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Villena, C.; Viana, R.; Bonet, J.; Garcia-Gimeno, M.A.; Casado, M.; Heredia, M.; Sanz, P. Astrocytes: New Players in Progressive Myoclonus Epilepsy of Lafora Type. Hum. Mol. Genet. 2018, 27, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brushia, R.J.; Walsh, D.A. Phosphorylase Kinase: The Complexity of Its Regulation Is Reflected in the Complexity of Its Structure. Front. Biosci. J. Virtual Libr. 1999, 4, D618–D641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome Sequencing Identifies the Cause of a Mendelian Disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.A.; Bhide, S.; Chin, E.; Ng, B.G.; Rhodenizer, D.; Zhang, V.W.; Sun, J.J.; Tanner, A.; Freeze, H.H.; Hegde, M.R. Targeted Polymerase Chain Reaction-Based Enrichment and next Generation Sequencing for Diagnostic Testing of Congenital Disorders of Glycosylation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, A.U.; Morell, R.J.; Belyantseva, I.A.; Khan, S.Y.; Boger, E.T.; Shahzad, M.; Ahmed, Z.M.; Riazuddin, S.; Khan, S.N.; Riazuddin, S.; et al. Targeted Capture and Next-Generation Sequencing Identifies C9orf75, Encoding Taperin, as the Mutated Gene in Nonsyndromic Deafness DFNB79. Am. J. Hum. Genet. 2010, 86, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cui, H.; Lee, N.-C.; Hwu, W.-L.; Chien, Y.-H.; Craigen, W.J.; Wong, L.-J.; Zhang, V.W. Clinical Application of Massively Parallel Sequencing in the Molecular Diagnosis of Glycogen Storage Diseases of Genetically Heterogeneous Origin. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Gailite, L.; Valenzuela-Palomo, A.; Sanoguera-Miralles, L.; Rots, D.; Kreile, M.; Velasco, E.A. UGT1A1 Variants c.864+5G>T and c.996+2_996+5del of a Crigler-Najjar Patient Induce Aberrant Splicing in Minigene Assays. Front. Genet. 2020, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Chui, M.H.; Yang, C.; Mehta, N.; Rai, V.; Zehir, A.; Momeni Boroujeni, A.; Ladanyi, M.; Mandelker, D. Somatic Intronic TP53 c.375+5G Mutations Are a Recurrent but under-Recognized Mode of TP53 Inactivation. J. Pathol. Clin. Res. 2022, 8, 14–18. [Google Scholar] [CrossRef]
- Ornaghi, A.P.; Meireles, M.R.; Botton, M.R.; Salzano, F.M.; Bandinelli, E.; Matte, U. Variants p.Pro2063Ser and p.Arg324* Co-Segregate in Type 3 von Willebrand Disease Patients from Southern Brazil. Haemoph. Off. J. World Fed. Hemoph. 2021, 27, e204–e213. [Google Scholar] [CrossRef]
- Piñero, T.A.; Soukarieh, O.; Rolain, M.; Alvarez, K.; López-Köstner, F.; Torrezan, G.T.; Carraro, D.M.; De Oliveira Nascimento, I.L.; Bomfim, T.F.; Machado-Lopes, T.M.B.; et al. MLH1 Intronic Variants Mapping to + 5 Position of Splice Donor Sites Lead to Deleterious Effects on RNA Splicing. Fam. Cancer 2020, 19, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, J.; Takada, S.; van Dijk, T.; Peña, F.; Boogaard, M.W.; van Duyvenvoorde, H.A.; Pico-Knijnenburg, I.; Wit, J.M.; van der Burg, M.; Mericq, V.; et al. Primary Ovarian Failure in Addition to Classical Clinical Features of Coats Plus Syndrome in a Female Carrying 2 Truncating Variants of CTC1. Horm. Res. Paediatr. 2021, 94, 448–455. [Google Scholar] [CrossRef]
- Carrière, C.; Mornon, J.-P.; Venien-Bryan, C.; Boisset, N.; Callebaut, I. Calcineurin B-like Domains in the Large Regulatory Alpha/Beta Subunits of Phosphorylase Kinase. Proteins 2008, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, O.W.; Traxler, K.W.; Fee, L.R.; Baldwin, B.A.; Carlson, G.M. Activators of Phosphorylase Kinase Alter the Cross-Linking of Its Catalytic Subunit to the C-Terminal One-Sixth of Its Regulatory Alpha Subunit. Biochemistry 1999, 38, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.R.; Malencik, D.A.; Johnson, C.M.; Carr, S.A.; Roberts, G.D.; Byles, C.A.; Anderson, S.R.; Heilmeyer, L.M.; Fischer, E.H.; Crabb, J.W. Purification and Characterization of Catalytic Fragments of Phosphorylase Kinase Gamma Subunit Missing a Calmodulin-Binding Domain. J. Biol. Chem. 1990, 265, 11740–11745. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Anwar, S.; Almatroudi, A.; Khan, A.A.; Alrumaihi, F.; Alsahli, M.A.; Rahmani, A.H. Hepatoprotective Effects of Garlic Extract against Carbon Tetrachloride (CCl4)-Induced Liver Injury via Modulation of Antioxidant, Anti-Inflammatory Activities and Hepatocyte Architecture. Appl. Sci. 2020, 10, 6200. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Ballester, M.; Sentandreu, E.; Luongo, G.; Santamaria, R.; Bolonio, M.; Alcoriza-Balaguer, M.I.; Palomino-Schätzlein, M.; Pineda-Lucena, A.; Castell, J.; Lahoz, A.; et al. Glutamine/Glutamate Metabolism Rewiring in Reprogrammed Human Hepatocyte-like Cells. Sci. Rep. 2019, 9, 17978. [Google Scholar] [CrossRef]
- Kulozik, P.; Jones, A.; Mattijssen, F.; Rose, A.J.; Reimann, A.; Strzoda, D.; Kleinsorg, S.; Raupp, C.; Kleinschmidt, J.; Müller-Decker, K.; et al. Hepatic Deficiency in Transcriptional Cofactor TBL1 Promotes Liver Steatosis and Hypertriglyceridemia. Cell Metab. 2011, 13, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Pashos, E.E.; Park, Y.; Wang, X.; Raghavan, A.; Yang, W.; Abbey, D.; Peters, D.T.; Arbelaez, J.; Hernandez, M.; Kuperwasser, N.; et al. Large, Diverse Population Cohorts of HiPSCs and Derived Hepatocyte-like Cells Reveal Functional Genetic Variation at Blood Lipid-Associated Loci. Cell Stem Cell 2017, 20, 558–570.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, J.L.; Duncan, S.A. IPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Llorens, G.; Lopez-Navarro, S.; Jaijo, T.; Castell, J.V.; Bort, R. Modeling a Novel Variant of Glycogenosis IXa Using a Clonal Inducible Reprogramming System to Generate “Diseased” Hepatocytes for Accurate Diagnosis. J. Pers. Med. 2022, 12, 1111. https://doi.org/10.3390/jpm12071111
Garcia-Llorens G, Lopez-Navarro S, Jaijo T, Castell JV, Bort R. Modeling a Novel Variant of Glycogenosis IXa Using a Clonal Inducible Reprogramming System to Generate “Diseased” Hepatocytes for Accurate Diagnosis. Journal of Personalized Medicine. 2022; 12(7):1111. https://doi.org/10.3390/jpm12071111
Chicago/Turabian StyleGarcia-Llorens, Guillem, Sergi Lopez-Navarro, Teresa Jaijo, Jose V. Castell, and Roque Bort. 2022. "Modeling a Novel Variant of Glycogenosis IXa Using a Clonal Inducible Reprogramming System to Generate “Diseased” Hepatocytes for Accurate Diagnosis" Journal of Personalized Medicine 12, no. 7: 1111. https://doi.org/10.3390/jpm12071111
APA StyleGarcia-Llorens, G., Lopez-Navarro, S., Jaijo, T., Castell, J. V., & Bort, R. (2022). Modeling a Novel Variant of Glycogenosis IXa Using a Clonal Inducible Reprogramming System to Generate “Diseased” Hepatocytes for Accurate Diagnosis. Journal of Personalized Medicine, 12(7), 1111. https://doi.org/10.3390/jpm12071111