
Fabry Disease in Slovakia: How the Situation Has Changed over 20 Years of Treatment

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
4.1. Fabry Disease in Females
4.2. Genetic Variants of Unknown Significance (GVUS)
4.3. Treatment of Fabry Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vieitez, I.; Souto-Rodriguez, O.; Fernandez-Mosquera, L.; San Millan, B.; Teijeira, S.; Fernandez-Martin, J.; Martinez-Sanchez, F.; Aldamiz-Echevarria, L.J.; Lopez-Rodriguez, M.; Navarro, C.; et al. Fabry disease in the Spanish population: Observational study with detection of 77 patients. Orphanet J. Rare Dis. 2018, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.; Müntze, J.; Gernert, J.; Spingler, L.; Nordbeck, P.; Wanner, C. Hot topics in Fabry disease. Postgrad. Med. J. 2018, 94, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linthorst, G.E.; Bouwman, M.G.; Wijburg, F.A.; Aerts, J.M.F.G.; Poorthuis, B.J.H.M.; Hollak, C.E.M. Screening for Fabry disease in high-risk populations: A systematic review. J. Med. Genet. 2010, 47, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sirrs, S.; Hollak, C.; Merkel, M.; Sechi, A.; Glamuzina, E.; Janssen, M.C.; Lachmann, R.; Langendonk, J.; Scarpelli, M.; Ben Omran, T.; et al. The Frequencies of Different Inborn Errors of Metabolism in Adult Metabolic Centres: Report from the SSIEM Adult Metabolic Physicians Group. JIMD Rep. 2015, 27, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. JASN 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Germain, D.P.; Arad, M.; Burlina, A.; Elliott, P.M.; Falissard, B.; Feldt-Rasmussen, U.; Hilz, M.J.; Hughes, D.A.; Ortiz, A.; Wanner, C.; et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease—A systematic literature review by a European panel of experts. Mol. Genet. Metab. 2019, 126, 224–235. [Google Scholar] [CrossRef]
- Hughes, D.A.; Aguiar, P.; Deegan, P.B.; Ezgu, F.; Frustaci, A.; Lidove, O.; Linhart, A.; Lubanda, J.-C.; Moon, J.C.; Nicholls, K.; et al. Early indicators of disease progression in Fabry disease that may indicate the need for disease-specific treatment initiation: Findings from the opinion-based PREDICT-FD modified Delphi consensus initiative. BMJ Open 2020, 10, e035182. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.; Mechtler, T.P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M.J.; Kasper, D.C. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease. Mol. Genet. Metab. 2018, 123, 148–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Hwu, W.-L.; Chien, Y.-H.; Lee, N.-C.; Chiang, S.-C.; Dobrovolny, R.; Huang, A.-C.; Yeh, H.-Y.; Chao, M.-C.; Lin, S.-J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef]
- Statistical Office of the Slovak Republic. Available online: https://slovak.statistics.sk/ (accessed on 8 April 2022).
- Sheng, S.; Wu, L.; Nalleballe, K.; Sharma, R.; Brown, A.; Ranabothu, S.; Kapoor, N.; Onteddu, S. Fabry’s disease and stroke: Effectiveness of enzyme replacement therapy (ERT) in stroke prevention, a review with meta-analysis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2019, 65, 83–86. [Google Scholar] [CrossRef]
- Reková, P.; Sedláková, K.; Dostálová, G.; Linhart, A. Fabryho choroba, přehled problematiky a nejčastější neurologické projevy. Čes. Slov. Neurol. Neurochir. 2018, 81, 156–163. [Google Scholar] [CrossRef]
- Nowicki, M.; Bazan-Socha, S.; Błażejewska-Hyzorek, B.; Gellert, R.; Imiela, J.; Kaźmierczak, J.; Kłopotowski, M.; Oko-Sarnowska, Z.; Pawlaczyk, K.; Ponikowski, P.; et al. Enzyme replacement therapy in Fabry disease in Poland: A position statement. Pol. Arch. Intern. Med. 2020, 130, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef]
- Reková, P.; Dostálová, G.; Kemlink, D.; Paulasová Schwabová, J.; Dubská, Z.; Vaneckova, M.; Mašek, M.; Kodet, O.; Poupětová, H.; Mazurová, S.; et al. Detailed Phenotype of GLA Variants Identified by the Nationwide Neurological Screening of Stroke Patients in the Czech Republic. J. Clin. Med. 2021, 10, 3543. [Google Scholar] [CrossRef] [PubMed]
- Holy, R.; Hlozkova, T.; Prochazkova, K.; Kalfert, D.; Hybnerova, F.; Ebelova, D.; Streubel, B.; Chovanec, M.; Gal, B.; Linhart, A.; et al. Prevalence of Fabry disease in men with tinnitus and sensorineural hearing loss. J. Appl. Biomed. 2021, 19, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Palecek, T.; Honzikova, J.; Poupetova, H.; Vlaskova, H.; Kuchynka, P.; Golan, L.; Magage, S.; Linhart, A. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: Prospective Fabry cardiomyopathy screening study (FACSS). J. Inherit. Metab. Dis. 2014, 37, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://datacube.statistics.sk/ (accessed on 8 April 2022).
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Wittmann, J.; Karg, E.; Turi, S.; Legnini, E.; Wittmann, G.; Giese, A.-K.; Lukas, J.; Gölnitz, U.; Klingenhäger, M.; Bodamer, O. Newborn screening for lysosomal storage disorders in hungary. JIMD Rep. 2012, 6, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Tai, C.-L.; Liu, M.-Y.; Yu, H.-C.; Chiang, C.-C.; Chiang, H.; Suen, J.-H.; Kao, S.-M.; Huang, Y.-H.; Wu, T.J.-T.; Yang, C.-F.; et al. The use of high resolution melting analysis to detect Fabry mutations in heterozygous females via dry bloodspots. Clin. Chim. Acta 2012, 413, 422–427. [Google Scholar] [CrossRef]
- Ferri, L.; Malesci, D.; Fioravanti, A.; Bagordo, G.; Filippini, A.; Ficcadenti, A.; Manna, R.; Antuzzi, D.; Verrecchia, E.; Donati, I.; et al. Functional and pharmacological evaluation of novel GLA variants in Fabry disease identifies six (two de novo) causative mutations and two amenable variants to the chaperone DGJ. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 481, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Shabbeer, J.; Yasuda, M.; Luca, E.; Desnick, R.J. Fabry disease: 45 novel mutations in the α-galactosidase A gene causing the classical phenotype. Mol. Genet. Metab. 2002, 76, 23–30. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Zhao, N.; Xiong, Q.; Zhou, Y.-A.; Wu, C.; Xiao, H. A Novel α-Galactosidase A Splicing Mutation Predisposes to Fabry Disease. Front. Genet. 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Lyon, M.F. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Monk, M.; Boubelik, M.; Lehnert, S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Dev. Camb. Engl. 1987, 99, 371–382. [Google Scholar] [CrossRef]
- Shvetsova, E.; Sofronova, A.; Monajemi, R.; Gagalova, K.; Draisma, H.H.M.; White, S.J.; Santen, G.W.E.; Chuva de Sousa Lopes, S.M.; Heijmans, B.T.; van Meurs, J.; et al. Skewed X-inactivation is common in the general female population. Eur. J. Hum. Genet. 2019, 27, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, C.M.; Ashley, G.A.; Burgert, T.S.; Enriquez, A.L.; D’Souza, M.; Desnick, R.J. Fabry disease: Thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol. Med. Camb. Mass 1997, 3, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nance, C.S.; Klein, C.J.; Banikazemi, M.; Dikman, S.H.; Phelps, R.G.; McArthur, J.C.; Rodriguez, M.; Desnick, R.J. Later-onset Fabry disease: An adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 2006, 63, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Monserrat, L.; Gimeno-Blanes, J.R.; Marín, F.; Hermida-Prieto, M.; García-Honrubia, A.; Pérez, I.; Fernández, X.; de Nicolas, R.; de la Morena, G.; Payá, E.; et al. Prevalence of Fabry Disease in a Cohort of 508 Unrelated Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 2399–2403. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M.F.G. Is it Fabry disease? Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 1181–1185. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P.; Oliveira, J.P.; Bichet, D.G.; Yoo, H.-W.; Hopkin, R.J.; Lemay, R.; Politei, J.; Wanner, C.; Wilcox, W.R.; Warnock, D.G. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispecialty Fabry disease genotype-phenotype workgroup. J. Med. Genet. 2020, 57, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortés, M.; Gallegos-Villalobos, Á.; Garcia, D.; García-Robles, J.A.; et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Martinez, D.; Aguiar, P.; Auray-Blais, C.; Beck, M.; Bichet, D.G.; Burlina, A.; Cole, D.; Elliott, P.; Feldt-Rasmussen, U.; Feriozzi, S.; et al. Standardising clinical outcomes measures for adult clinical trials in Fabry disease: A global Delphi consensus. Mol. Genet. Metab. 2021, 132, 234–243. [Google Scholar] [CrossRef]
- Shen, J.-S.; Busch, A.; Day, T.S.; Meng, X.-L.; Yu, C.I.; Dabrowska-Schlepp, P.; Fode, B.; Niederkrüger, H.; Forni, S.; Chen, S.; et al. Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J. Inherit. Metab. Dis. 2016, 39, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef]
- Ashe, K.M.; Budman, E.; Bangari, D.S.; Siegel, C.S.; Nietupski, J.B.; Wang, B.; Desnick, R.J.; Scheule, R.K.; Leonard, J.P.; Cheng, S.H.; et al. Efficacy of Enzyme and Substrate Reduction Therapy with a Novel Antagonist of Glucosylceramide Synthase for Fabry Disease. Mol. Med. Camb. Mass 2015, 21, 389–399. [Google Scholar] [CrossRef]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J.; International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Beck, M.; Hughes, D.; Kampmann, C.; Larroque, S.; Mehta, A.; Pintos-Morell, G.; Ramaswami, U.; West, M.; Wijatyk, A.; Giugliani, R. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis. Mol. Genet. Metab. Rep. 2015, 3, 21–27. [Google Scholar] [CrossRef]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Arngrímsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: The European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- GALAFOLD (Migalastat) Capsules, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208623lbl.pdf (accessed on 13 April 2022).
- Galafold: Annex I-Summary of Product Characteristics. Last Update 25 February 2022. Available online: https://www.ema.europa.eu/en/documents/product-information/galafold-epar-product-information_en.pdf (accessed on 13 April 2022).
- Parini, R.; Pintos-Morell, G.; Hennermann, J.B.; Hsu, T.-R.; Karabul, N.; Kalampoki, V.; Gurevich, A.; Ramaswami, U. Analysis of Renal and Cardiac Outcomes in Male Participants in the Fabry Outcome Survey Starting Agalsidase Alfa Enzyme Replacement Therapy Before and After 18 Years of Age. Drug Des. Devel. Ther. 2020, 14, 2149–2158. [Google Scholar] [CrossRef]
- Wanner, C.; Arad, M.; Baron, R.; Burlina, A.; Elliott, P.M.; Feldt-Rasmussen, U.; Fomin, V.V.; Germain, D.P.; Hughes, D.A.; Jovanovic, A.; et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol. Genet. Metab. 2018, 124, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Hopkin, R.J.; Jefferies, J.L.; Laney, D.A.; Lawson, V.H.; Mauer, M.; Taylor, M.R.; Wilcox, W.R.; Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol. Genet. Metab. 2016, 117, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Baig, S.; Abdel-Gadir, A.; Medina-Menacho, K.; Rosmini, S.; Captur, G.; Tchan, M.; Geberhiwot, T.; Murphy, E.; et al. Cardiac Phenotype of Prehypertrophic Fabry Disease. Circ. Cardiovasc. Imaging 2018, 11, e007168. [Google Scholar] [CrossRef] [Green Version]
- Nordin, S.; Kozor, R.; Vijapurapu, R.; Augusto, J.B.; Knott, K.D.; Captur, G.; Treibel, T.A.; Ramaswami, U.; Tchan, M.; Geberhiwot, T.; et al. Myocardial Storage, Inflammation, and Cardiac Phenotype in Fabry Disease After One Year of Enzyme Replacement Therapy. Circ. Cardiovasc. Imaging 2019, 12, e009430. [Google Scholar] [CrossRef]
- Camporeale, A.; Pieroni, M.; Pieruzzi, F.; Lusardi, P.; Pica, S.; Spada, M.; Mignani, R.; Burlina, A.; Bandera, F.; Guazzi, M.; et al. Predictors of Clinical Evolution in Prehypertrophic Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e008424. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry disease—A multisystemic disease with gastrointestinal manifestations. Gut Microbes 2022, 14, 2027852. [Google Scholar] [CrossRef]
- Juríčková, K.; Mattošová, S.; Šalingová, A.; Jungová, P.; Brennerová, K.; Kolníková, M.; Košťálová, Ľ.; Hlavatá, A. Lyzozómové choroby—Vývoj diagnostiky a liečby na Slovensku. Čes.-Slov. Pediatr. 2018, 73, 408–416. [Google Scholar]
- Juríčková, K.; Hlavatá, A.; Špalek, P.; Jungová, P.; Kolníková, M. Život z pohľadu dospelého pacienta s lyzozómovým ochorením. In 31. Pracovné Dni Dedičné Metabolické Poruchy; Meritis Praha: Bratislava, Slovakia, 2016; pp. 26–27. [Google Scholar]
- Nowicki, M.; Bazan-Socha, S.; Kłopotowski, M.; Błażejewska-Hyżorek, B.; Kusztal, M.; Pawlaczyk, K.; Sławek, J.; Oko, A.; Oko-Sarnowska, Z. Considerations for Home-Based Treatment of Fabry Disease in Poland during the COVID-19 Pandemic and Beyond. Int. J. Environ. Res. Public. Health 2021, 18, 8242. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr. | GLA Variant | Sex | Age at Diagnosis (Years) | Phenotype Previously Described | Type of Screening | Therapy | Age at Treatment (Years) | Comment |
|---|---|---|---|---|---|---|---|---|
| Patient with pathogenic variant | ||||||||
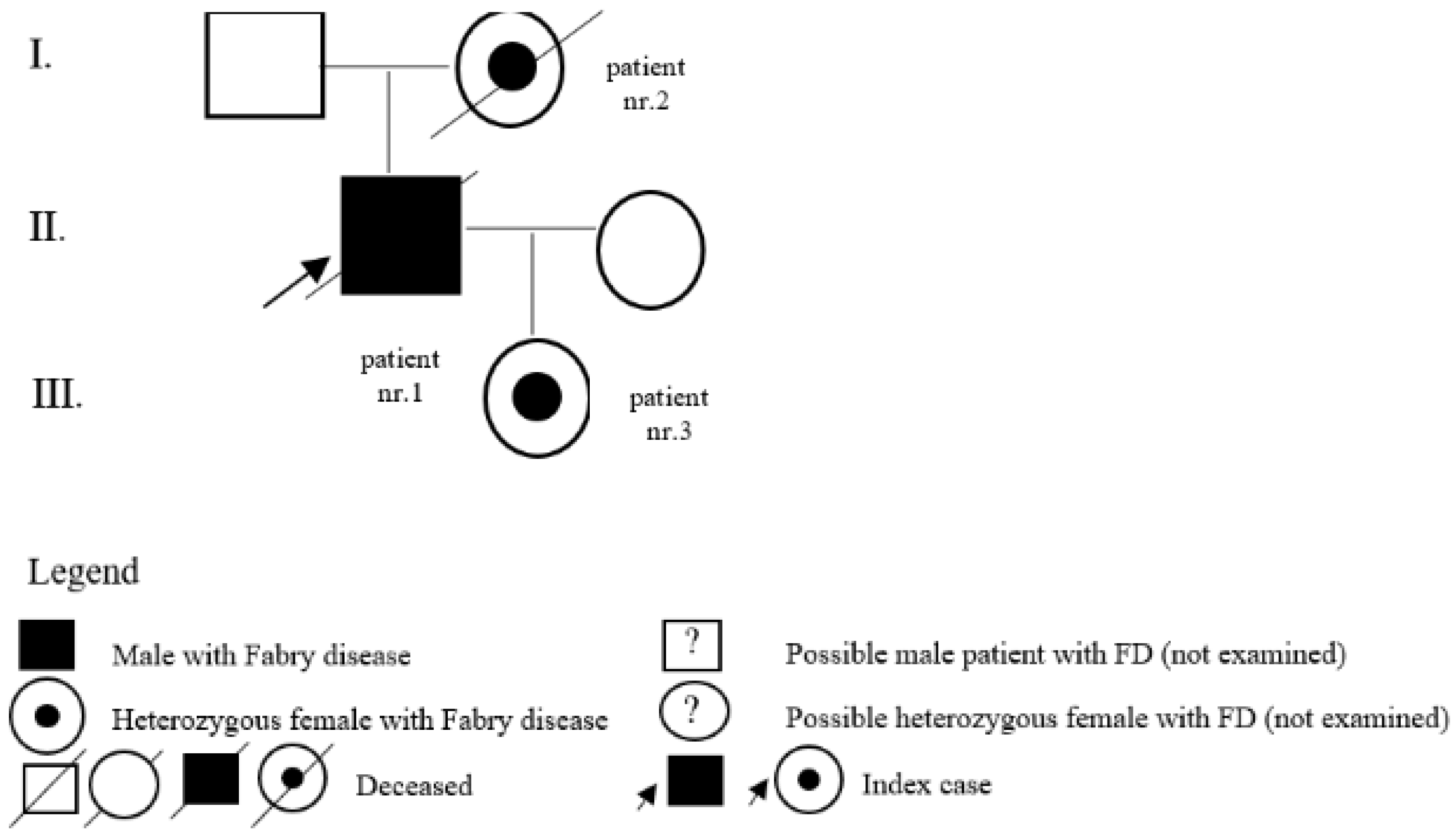
| 1 | c.128G>A (p.Gly43Asp) | M | 20 | classic | kidney | Y | 24 | son of patient nr. 2 and father of patient nr. 3 |
| 2 | c.128G>A (p.Gly43Asp) | F | 50 | classic | pedigree | Y | 62 | mother of patient nr. 1 |
| 3 | c.128G>A (p.Gly43Asp) | F | 0 | classic | pedigree | N | daughter of patient nr. 1 | |
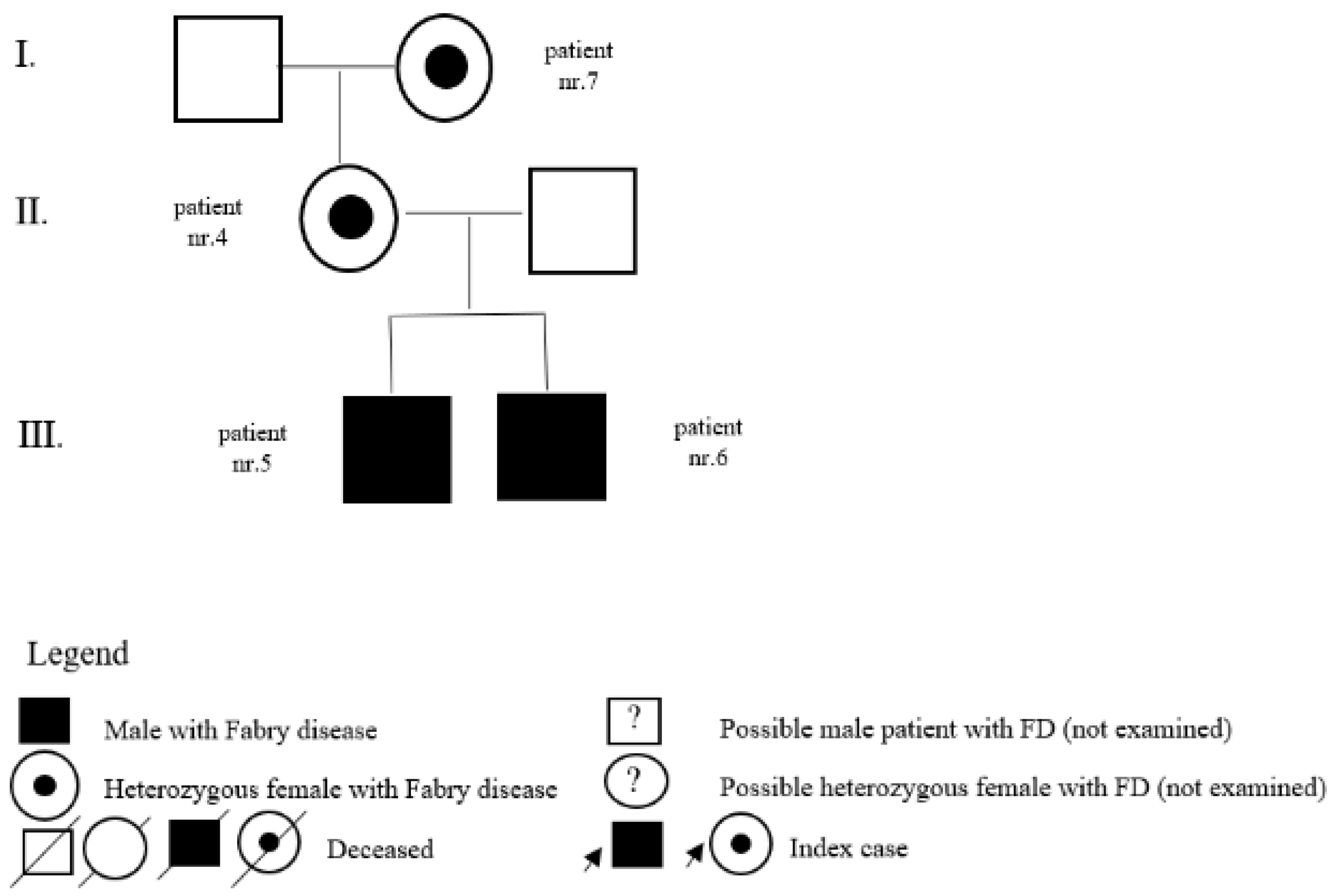
| 4 | c.463G>C (p.D155H) | F | 23 | classic | pedigree | N | mother of patients nr. 5 and nr. 6, daughter of patient nr. 7 | |
| 5 | c.463G>C (p.D155H) | M | 4 | classic | pedigree | Y | 18 | brother of patient nr. 6 |
| 6 | c.463G>C (p.D155H) | M | 5 | classic | pedigree | Y | 13 | brother of patient nr. 5 |
| 7 | c.463G>C (p.D155H) | F | 52 | classic | pedigree | N | mother of patient nr. 4 | |
| 8 | NA | M | 29 | NA | NA | |||
| 9 | NA | M | 35 | NA | NA | Y | 35 | |
| 10 | c.736delA (p.Thr246HisfsTer23) | M | 25 | undescribed/classic | kidney | Y | 25 | son of patient nr. 11 and father of patient nr. 12 |
| 11 | c.736delA (p.Thr246HisfsTer23) | F | 59 | undescribed/classic | pedigree | N | mother of patient nr. 10 | |
| 12 | c.736delA (p.Thr246HisfsTer23) | F | 0 | undescribed/classic | pedigree | N | daughter of patient nr. 10 | |
| 13 | c.169C>T (p.Q57X) | M | 54 | classic | LVH | Y | 54 | father of patient nr. 14 |
| 14 | c.169C>T (p.Q57X) | F | 24 | classic | pedigree | N | daughter of patient nr. 13 | |
| 15 | c.98A>G (p.D33G) | M | 58 | classic | LVH | N | father of patients nr. 18 and nr. 19, grandfather of patients nr. 16 and nr. 17 | |
| 16 | c.98A>G (p.D33G) | M | 8 | classic | pedigree | Y | 14 | son of patient nr. 19 and brother of patient nr. 17 |
| 17 | c.98A>G (p.D33G) | F | 15 | classic | pedigree | N | daughter of patient nr. 19 and sister of patient nr. 16 | |
| 18 | c.98A>G (p.D33G) | F | 31 | classic | pedigree | N | daughter of patient nr. 15 | |
| 19 | c.98A>G (p.D33G) | F | 33 | classic | pedigree | N | daughter of patient nr. 15, mother of patients nr. 16 and nr. 17 | |
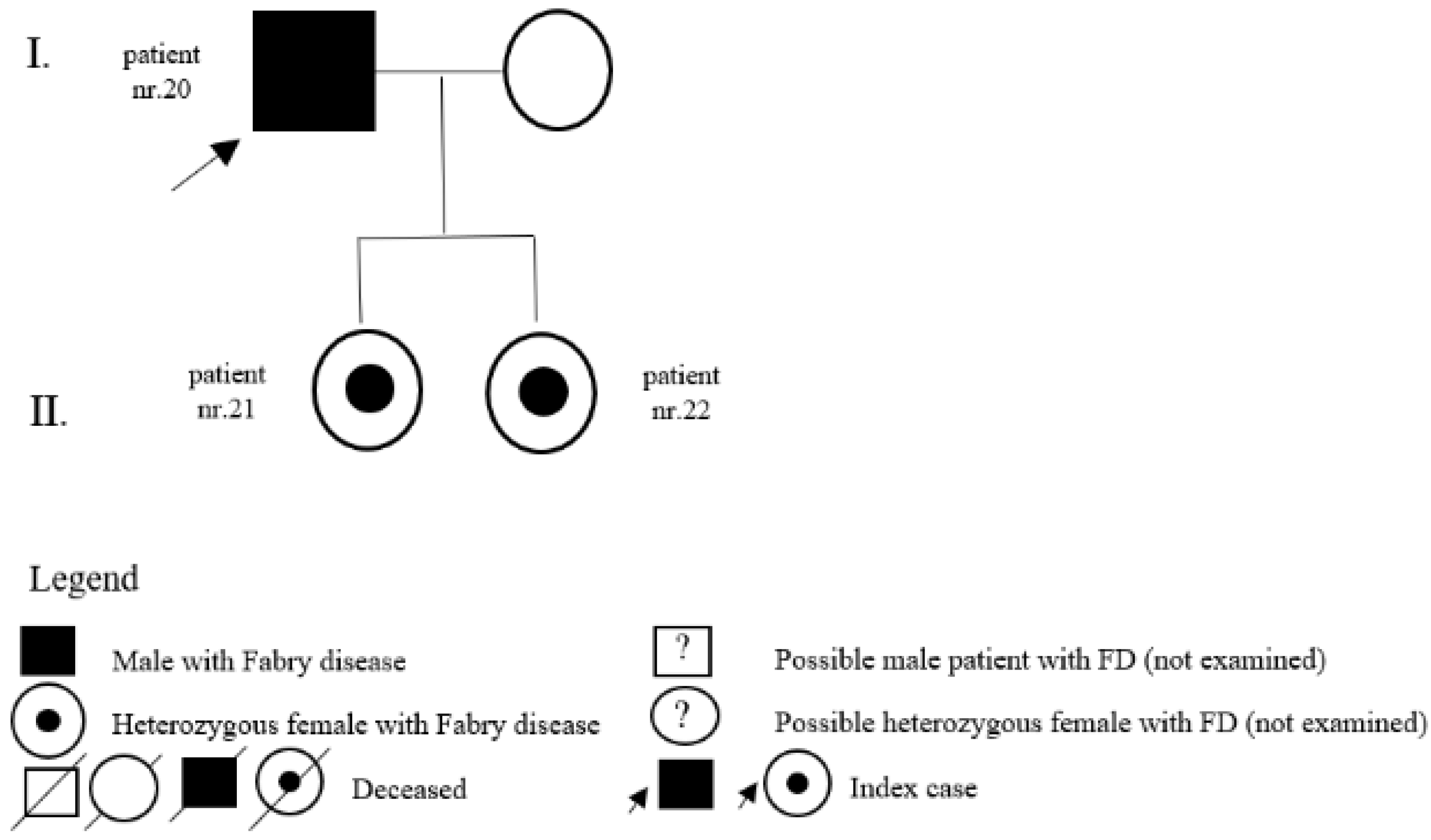
| 20 | c.334C>T (p.R112C) | M | 35 | classic | LVH | Y | 35 | father of patients nr. 21 and nr. 22 |
| 21 | c.334C>T (p.R112C) | F | 0 | classic | pedigree | N | daughter of patient nr. 20 | |
| 22 | c.334C>T (p.R112C) | F | 0 | classic | pedigree | N | daughter of patient nr. 20 | |
| 23 | c.334C>T (p.R112C) | M | 38 | classic | LVH | Y | 38 | |
| 24 | c.730G>A (p.Asp244Asn) | F | 50 | classic | LVH | Y | 51 | |
| 25 | c.1073A>G (p.Glu358Gly) | F | 85 | classic | LVH | N | ||
| 26 | c.801+1G>A, (p.L268IfsX3) | M | 55 | renal | LVH | Y | 56 | new description of phenotype—arrythmia, LVH, depression |
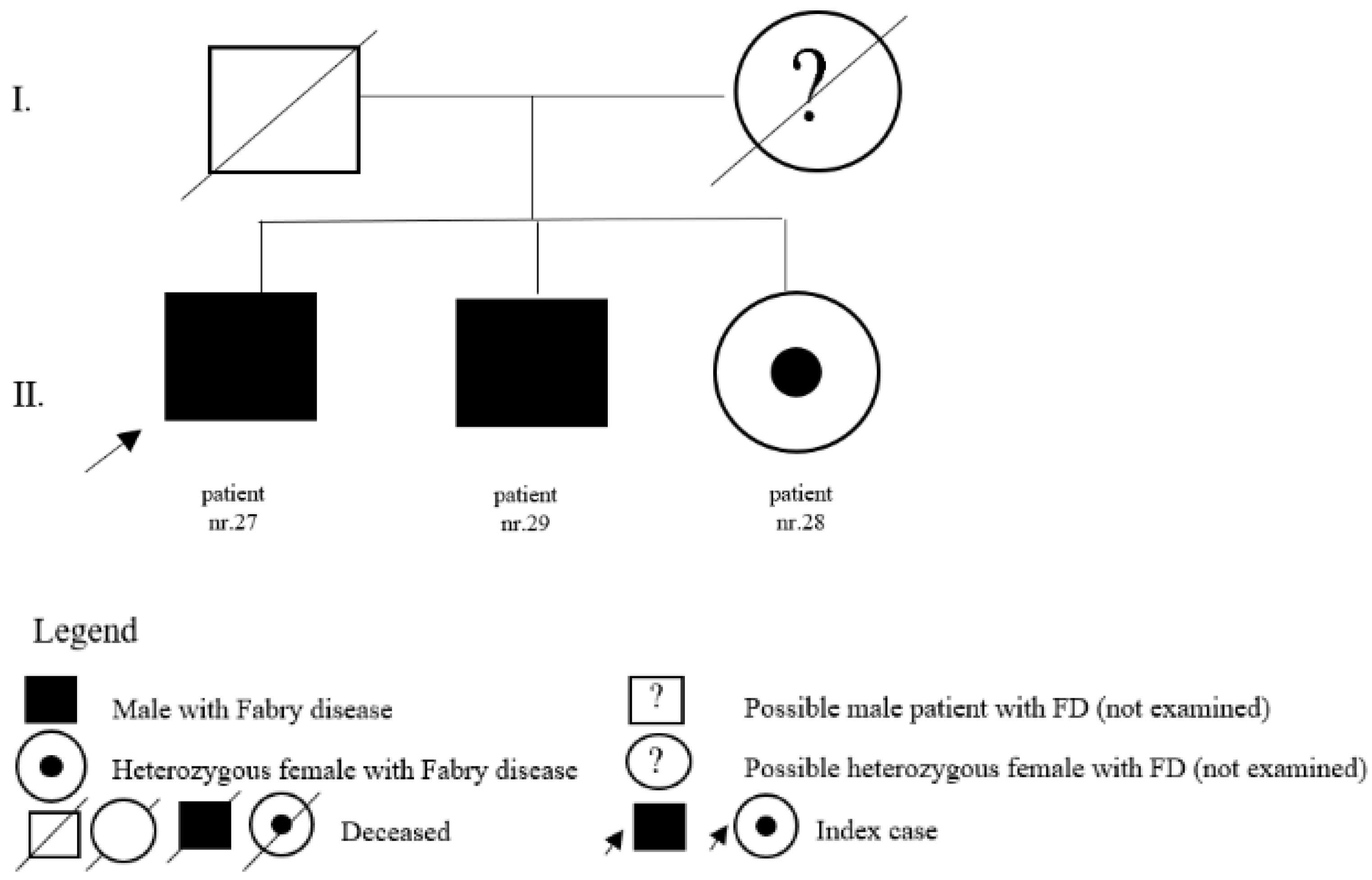
| 27 | c.758T>C (p.Ile753Thr) | M | 65 | classic | LVH | Y | 65 | brother of patients nr. 28 and nr. 29 |
| 28 | c.758T>C (p.Ile753Thr) | F | 55 | classic | pedigree | N | sister of patient nr. 27 and nr. 29 | |
| 29 | c.758T>C (p.Ile753Thr) | M | 62 | classic | pedigree | N | brother of patients nr. 27 and nr. 28 | |
| 30 | c.157 A>C (p.Asn53His) | F | 42 | undescribed | SM | Y | 43 | arrythmia, neurological impairment |
| 31 | c.203T>C (p.Leu68Pro) | F | 46 | undescribed | LVH | N | hypertrophic cardiomyopathy | |
| Data of 48 Slovak FD patients—part 2 | ||||||||
| Patients with GVUS | ||||||||
| 32 | c.352C>T (p.R118C also p.Arg118Cys) | F | 75 | kidney | N | mother of patient nr.33 | ||
| 33 | c.352C>T (p.R118C also p.Arg118Cys) | M | 45 | pedigree | N | son of patient nr. 32 | ||
| 34 | c.352C>T (p.R118C also p.Arg118Cys) | M | 41 | kidney | N | |||
| 35 | c.427G>A(p.A143T also p.Ala143Thr) | F | 73 | kidney | N | sister of patient nr. 36 | ||
| 36 | c.427G>A(p.A143T also p.Ala143Thr) | M | 78 | pedigree | N | brother of patient nr. 35 | ||
| 37 | c.427G>A(p.A143T also p.Ala143Thr) | F | 43 | pedigree | N | daughter of patient nr. 36 | ||
| 38 | c.427G>A(p.A143T also p.Ala143Thr) | F | 46 | pedigree | N | daughter of patient nr. 36 | ||
| 39 | c.427G>A(p.A143T also p.Ala143Thr) | F | 49 | pedigree | N | daughter of patient nr. 36 | ||
| 40 | c.427G>A(p.A143T also p.Ala143Thr) | M | 14 | pedigree | N | son of patient nr. 37 | ||
| 41 | c.427G>A(p.A143T also p.Ala143Thr) | M | 62 | pedigree | N | brother of patient nr. 35 | ||
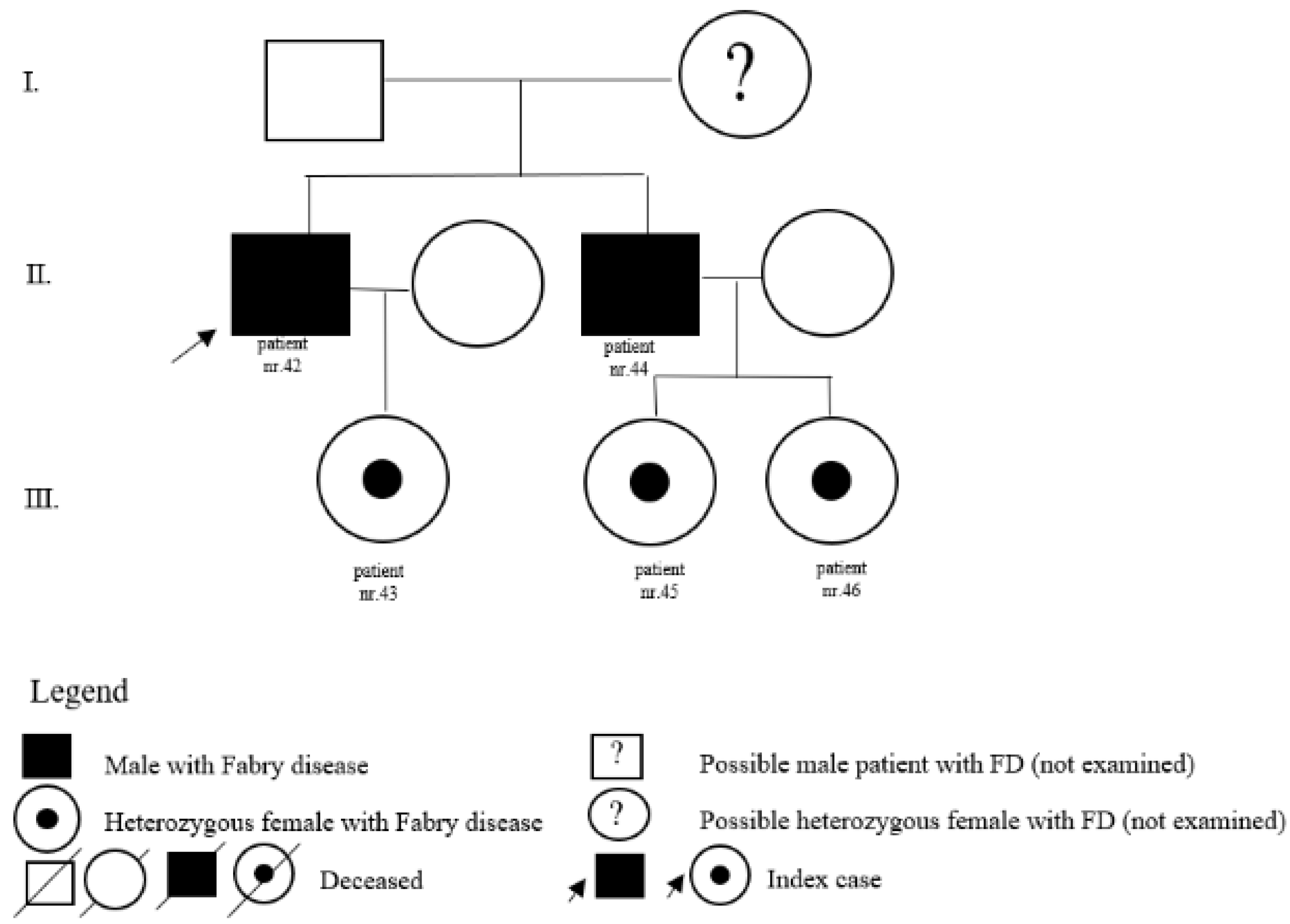
| 42 | c.427G>A(p.A143T also p.Ala143Thr) | M | 73 | kidney | N | father of patient nr. 43 and brother of patient nr. 44 | ||
| 43 | c.427G>A(p.A143T also p.Ala143Thr) | F | 43 | pedigree | N | daughter of patient nr. 42 | ||
| 44 | c.427G>A(p.A143T also p.Ala143Thr) | M | 69 | pedigree | N | father of patients nr.45 and nr. 46 and brother of patient nr. 42 | ||
| 45 | c.427G>A(p.A143T also p.Ala143Thr) | F | 45 | pedigree | N | daughter of patient nr. 44 | ||
| 46 | c.427G>A(p.A143T also p.Ala143Thr) | F | 46 | pedigree | N | daughter of patient nr. 44 | ||
| 47 | c.427G>A(p.A143T also p.Ala143Thr) | F | 82 | kidney | N | |||
| 48 | c.427G>A(p.A143T also p.Ala143Thr) | F | 31 | SM | N | |||
| Nr. | Variant | Sex | Age at First Symptom (Years) | Age at Diagnosis (Years) | Phenotype Previously Described | Age at Treatment (Years) | Time from First Symptom to Diagnosis | Time from Diagnosis to Treatment | Therapy | Duration of ERT (Years) | Effect of Therapy | LysoGb3 Prior ERT | LysoGb3 after ERT (Last Value) | Age at Death (Years) and Reason |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.128G>A (p.Gly43Asp) | M | 14 | 20 | classic | 24 | 6 | 4 | ERT (agalsidase beta, agalsidase alfa) | 13 | stabilization of renal parameters; SWB improved | NA | NA | 37; ruptured brain aneurysm |
| 2 | c.128G>A (p.Gly43Asp) | F | 54 | 50 | classic | 62 | −4 | 12 | ERT (agalsidase alfa) | 7 | temporary improvement of CV parameters, improvement of physical activities | NA | 6.0 | 69; heart failure |
| 3 | c.463G>C (p.D155H) | M | 12 | 4 | classic | 18 | −8 | 14 | ERT (agalsidase beta) | 8 | stabilization and improvement of renal and CV parameters, SWB improved | NA | 7.9 (N 0–3.5) ng/mL | |
| 4 | c.463G>C (p.D155H) | M | 13 | 5 | classic | 13 | −7 | 8 | ERT (agalsidase beta) | 4 | renal and CV parameters in reference level; normal psychomotor development | 142.9 (N 0–3.5) ng/ml | 11.1 (N 0–3.5) ng/mL | |
| 5 | NA | M | 15 | 35 | NA | 35 | 20 | 0 | ERT (agalsidase beta) | NA | NA | NA | NA | |
| 6 | c.736delA (p.Thr246HisfsTer23) | M | 14 | 25 | undescribed/classic | 25 | 11 | 0 | ERT (agalsidase beta) | 16 | stabilization and improvement of renal parameters; pain relief; SWB improved | NA | 25.1 (N 0–3.5) ng/mL | |
| 7 | c.169C>T (p.Q57X) | M | 35 | 54 | classic | 54 | 19 | 0 | ERT (agalsidase alfa) | 8 | temporary improvement of CV parameters, stabilization of renal parameters, SWB improved | NA | 40 (N 0–1.8) ng/mL | 62; multiorgan failure after repeated stroke |
| 8 | c.98A>G (p.D33G) | M | 14 | 8 | classic | 14 | −6 | 6 | ERT (agalsidase alfa) | 2 | stabilization of renal parameters; normal psychomotor development | 38 (N 0–1.8) ng/ml | 11.7 (N 0–1.8) ng/mL | |
| 9 | c.334C>T (p.R112C) | M | 14 | 35 | classic | 35 | 21 | 0 | ERT (agalsidase alfa) | 9 | stabilization and improvement of renal and CV parameters; SWB improved | NA | 14.7 (N 0–3.5) ng/mL | |
| 10 | c.334C>T (p.R112C) | M | 10 | 38 | classic | 38 | 28 | 0 | ERT (agalsidase beta) | 3 | stabilization of renal and CV parameters, improvement of physical activities | 89.5 (N 0–3.5) ng/ml | 16.0 (N 0–3.5) ng/mL | |
| 11 | c.730G>A (p.Asp244Asn) | F | 16 | 50 | classic | 51 | 34 | 1 | ERT (agalsidase alfa) | 1 | stabilization and improvement of renal and CV parameters; SWB improved | 5.7 (N 0–1.8) ng/mL | 4.2 (N 0–1.8) ng/mL | |
| 12 | c.801+1G>A, (p.L268IfsX3) | M | 10 | 55 | renal | 56 | 45 | 1 | ERT (agalsidase alfa) | 3.5 | stabilization of renal parameters; end of ERT after 3.5 years, due to deterioration of cardiological findings | 95.8 (N 0–1.8) ng/mL | 48.1 (N 0–1.8) ng/mL | |
| 13 | c.758T>C (p.Ile753Thr) | M | 64 | 65 | classic | 65 | 1 | 0 | ERT (agalsidase beta) | 3 | stabilization of renal and improvement of CV parameters, improvement of physical activities | 18.1 (N 0–3.5) ng/mL | 8.6 (N 0–3.5) ng/mL | |
| 14 | c.157 A>C p.(Asn53His) | F | 16 | 42 | undescribed | 43 | 26 | 1 | ERT (agalsidase alfa) | 1 | stabilization of renal and CV parameters; improvement in gross motor function and self-activity | 1.3 (N 0–1.8) ng/mL | 1.0 (N 0–1.8) ng/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurickova, K.; Jungova, P.; Petrovic, R.; Mattosova, S.; Hlavata, T.; Kostalova, L.; Hlavata, A. Fabry Disease in Slovakia: How the Situation Has Changed over 20 Years of Treatment. J. Pers. Med. 2022, 12, 922. https://doi.org/10.3390/jpm12060922
Jurickova K, Jungova P, Petrovic R, Mattosova S, Hlavata T, Kostalova L, Hlavata A. Fabry Disease in Slovakia: How the Situation Has Changed over 20 Years of Treatment. Journal of Personalized Medicine. 2022; 12(6):922. https://doi.org/10.3390/jpm12060922
Chicago/Turabian StyleJurickova, Katarina, Petra Jungova, Robert Petrovic, Slavomira Mattosova, Tereza Hlavata, Ludmila Kostalova, and Anna Hlavata. 2022. "Fabry Disease in Slovakia: How the Situation Has Changed over 20 Years of Treatment" Journal of Personalized Medicine 12, no. 6: 922. https://doi.org/10.3390/jpm12060922
APA StyleJurickova, K., Jungova, P., Petrovic, R., Mattosova, S., Hlavata, T., Kostalova, L., & Hlavata, A. (2022). Fabry Disease in Slovakia: How the Situation Has Changed over 20 Years of Treatment. Journal of Personalized Medicine, 12(6), 922. https://doi.org/10.3390/jpm12060922